ZusammensetzungWirkstoffe
Odevixibat (als Odevixibat-Sesquihydrat).
Hilfsstoffe
Kapselinhalt
Mikrokristalline Cellulose, Hypromellose.
Kapselhülle
Bylvay 200 Mikrogramm und 600 Mikrogramm Hartkapseln:
Hypromellose, Titandioxid, Eisenoxid gelb.
Bylvay 400 Mikrogramm und 1200 Mikrogramm Hartkapseln:
Hypromellose, Titandioxid, Eisenoxid gelb, Eisenoxid rot.
Drucktinte
Schellack, Propylenglycol, Eisenoxid schwarz.
Darreichungsform und Wirkstoffmenge pro EinheitBylvay 200 µg Hartkapseln
Kapsel der Grösse 0 (21,7 mm × 7,64 mm) mit lichtundurchlässigem elfenbeinweissem Kapseloberteil und lichtundurchlässigem weissem Kapselunterteil mit der Aufschrift "A200" in schwarzer Tinte.
Jede Hartkapsel enthält Odevixibat-Sesquihydrat entsprechend 200 Mikrogramm Odevixibat.
Bylvay 400 µg Hartkapseln
Kapsel der Grösse 3 (15,9 mm × 5,82 mm) mit lichtundurchlässigem orangefarbenem Kapseloberteil und lichtundurchlässigem weissem Kapselunterteil mit der Aufschrift "A400" in schwarzer Tinte.
Jede Hartkapsel enthält Odevixibat-Sesquihydrat entsprechend 400 Mikrogramm Odevixibat.
Bylvay 600 µg Hartkapseln
Kapsel der Grösse 0 (21,7 mm × 7,64 mm) mit lichtundurchlässigem elfenbeinweissem Kapselober- und -unterteil mit der Aufschrift "A600" in schwarzer Tinte.
Jede Hartkapsel enthält Odevixibat-Sesquihydrat entsprechend 600 Mikrogramm Odevixibat.
Bylvay 1200 µg Hartkapseln
Kapsel der Grösse 3 (15,9 mm × 5,82 mm) mit lichtundurchlässigem orangefarbenem Kapselober- und -unterteil mit der Aufschrift "A1200" in schwarzer Tinte.
Jede Hartkapsel enthält Odevixibat-Sesquihydrat entsprechend 1200 Mikrogramm Odevixibat.
Indikationen/AnwendungsmöglichkeitenBylvay wird angewendet zur Behandlung von cholestatischem Pruritus bei Patienten mit
progressiver familiärer intrahepatischer Cholestase (PFIC) im Alter von 6 Monaten und älter;
-Alagille-Syndrom (ALGS) im Alter von 12 Monaten und älter.
Warnhinweise und Vorsichtsmassnamen sind zu berücksichtigen (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Klinische Wirksamkeit" ).
Dosierung/AnwendungDosierung bei PFIC
Die Behandlung muss von Ärzten eingeleitet und überwacht werden, die Erfahrung in der Behandlung der PFIC haben.
Die empfohlene Dosis von Bylvay beträgt 40 µg/kg und wird einmal täglich morgens oral verabreicht.
Dosiseskalation
Wenn nach 3-monatiger kontinuierlicher Therapie kein angemessenes klinisches Ansprechen (Linderung des Pruritus und Senkung des Gallensäurespiegels im Serum) erreicht wird, kann die Dosis auf 120 µg/kg/Tag erhöht werden, wobei die Tageshöchstdosis von 7200 µg nicht überschritten werden darf (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Eine alternative Behandlung sollte bei Patienten in Betracht gezogen werden, für die nach 6-monatiger kontinuierlicher täglicher Behandlung mit Bylvay kein therapeutischer Nutzen nachgewiesen werden kann.
Dosierung bei ALGS
Die Behandlung muss von Ärzten eingeleitet und überwacht werden, die Erfahrung in der Behandlung des ALGS haben.
Die empfohlene Dosis von Bylvay zur Behandlung des ALGS beträgt 120 µg/kg und wird einmal täglich morgens oral verabreicht. Eine alternative Behandlung sollte bei Patienten in Betracht gezogen werden, für die nach 6-monatiger kontinuierlicher täglicher Behandlung mit Bylvay kein therapeutischer Nutzen nachgewiesen werden kann.
Dosisreduktion
Bei Patienten mit ALGS kann eine Dosisreduktion auf 40 µg/kg/Tag erwogen werden, wenn Verträglichkeitsprobleme auftreten und keine anderen Ursachen vorliegen. Sobald sich die Verträglichkeitsprobleme stabilisiert haben, kann die Dosis auf 120 µg/kg/Tag erhöht werden.
Alle verfügbaren Kapselstärken sind austauschbar und können sowohl im Ganzen geschluckt als auch geöffnet und aufgestreut werden. Die Auswahl der Stärke zur Erzielung der Tagesgesamtdosis richtet sich nach der prognostizierten Anwenderfreundlichkeit für den jeweiligen Patienten, d.h. nach der Gesamtzahl der Kapseln, der Grösse der Kapseln und der Fähigkeit, ganze Kapseln zu schlucken.
Tabelle 1 zeigt die Stärke und Anzahl der Kapseln, die täglich entsprechend dem Körpergewicht verabreicht werden sollten, um eine Dosierung von ca. 40 µg/kg/Tag zu erreichen.
Tabelle 1: Anzahl der zum Erreichen der Nominaldosis von 40 µg/kg/Tag erforderlichen Bylvay-Kapseln
Körpergewicht (kg) Anzahl an 200-µg-Kapseln Anzahl an 400-µg-Kapseln
4 bis < 7,5 1 oder nicht zutreffend
7,5 bis < 12,5 2 oder 1
12,5 bis < 17,5 3 oder nicht zutreffend
17,5 bis < 25,5 4 oder 2
25,5 bis < 35,5 6 oder 3
35,5 bis < 45,5 8 oder 4
45,5 bis < 55,5 10 oder 5
≥55,5 12 oder 6
Die durch Fettdruck hervorgehobene Kapselstärke/-anzahl wird basierend auf der prognostizierten Anwenderfreundlichkeit empfohlen.
Tabelle 2 zeigt die Stärke und Anzahl der Kapseln, die basierend auf dem Körpergewicht täglich verabreicht werden sollten, um eine Dosierung von ungefähr 120 µg/kg/Tag zu erreichen, wobei die Tageshöchstdosis von 7200 µg/Tag nicht überschritten werden darf.
Tabelle 2: Anzahl der zum Erreichen der Nominaldosis von 120 µg/kg/Tag erforderlichen Bylvay-Kapseln
Körpergewicht (kg) Anzahl an 600-µg-Kapseln Anzahl an 1200-µg-Kapseln
4 bis < 7,5 1 oder nicht zutreffend
7,5 bis < 12,5 2 oder 1
12,5 bis < 17,5 3 oder nicht zutreffend
17,5 bis < 25,5 4 oder 2
25,5 bis < 35,5 6 oder 3
35,5 bis < 45,5 8 oder 4
45,5 bis < 55,5 10 oder 5
≥55,5 12 oder 6
Die durch Fettdruck hervorgehobene Kapselstärke/-anzahl wird basierend auf der prognostizierten Anwenderfreundlichkeit empfohlen.
Versäumte Dosen
Wenn eine Dosis Bylvay versäumt wird, sollte der Patient die versäumte Dosis innerhalb von 12 Stunden nach der üblichen Einnahmezeit einnehmen und anschliessend den ursprünglichen Dosierungsrhythmus wieder aufnehmen. Sind mehr als 12 Stunden nach der üblichen Einnahmezeit vergangen, soll diese Dosis übersprungen und der ursprüngliche Dosierungsrhythmus wiederaufgenommen werden.
Spezielle Dosierungsanweisungen
Patienten mit Nierenfunktionsstörung
Es liegen keine klinischen Daten zur Anwendung von Bylvay bei Patienten mit mittelschwerer bis schwerer Nierenfunktionsstörung oder terminaler dialysepflichtiger Niereninsuffizienz (ESRD) vor (siehe "Pharmakokinetik" ). Aufgrund der minimalen Plasmakonzentrationen und der vernachlässigbaren renalen Ausscheidung ist bei Patienten mit Nierenfunktionsstörung jedoch keine Dosisanpassung erforderlich.
Patienten mit Leberfunktionsstörung
Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe "Pharmakokinetik" ).
Bylvay wurde bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) nicht untersucht. Setzen Sie Bylvay dauerhaft ab, wenn Zeichen einer hepatischen Dekompensation auftreten (z.B. Varizenblutung, Aszites, hepatische Enzephalopathie; siehe "Kontraindikationen" und "Warnhinweise und Vorsichtsmassnahmen" ).
Ältere Patienten
Für Patienten ab 65 Jahren liegen keine klinischen Daten vor.
Pädiatrische Population
Bylvay ist für die Anwendung bei PFIC Patienten unter 6 Monaten und ALGS Patienten unter 12 Monaten nicht zugelassen.
Art der Anwendung
Bylvay wird per os eingenommen. Das Arzneimittel ist morgens mit oder ohne Nahrung einzunehmen.
Die grösseren Kapseln mit 200 µg und 600 µg sind dazu bestimmt, geöffnet und auf weiche Lebensmittel gestreut oder in eine Flüssigkeit gegeben zu werden, können aber auch im Ganzen geschluckt werden.
Die kleineren Kapseln mit 400 µg und 1200 µg sind dazu bestimmt, im Ganzen geschluckt zu werden, können aber auch geöffnet und auf weiche Lebensmittel gestreut oder in eine Flüssigkeit gegeben werden.
Wird die Kapsel im Ganzen geschluckt, sollte der Patient angewiesen werden‚ sie morgens mit einem Glas Wasser einzunehmen.
Einnahme zusammen mit Nahrung mit weicher Konsistenz
Beim Öffnen der Kapseln zur Verteilung des Inhalts auf Nahrung mit weicher Konsistenz sollte der Patient bzw. die Betreuungsperson angewiesen werden, wie folgt vorzugehen:
1.Eine kleine Menge (bis zu 30 ml/2 Esslöffel) weicher Nahrung (Joghurt, Apfelmus, Haferbrei, Bananenpüree, Karottenpüree, Schokoladenpudding oder Milchreis) in eine Schüssel geben. Die Temperatur der Nahrung sollte der Raumtemperatur entsprechen oder darunter liegen.
2.Die Kapsel horizontal an beiden Enden halten, in entgegengesetzte Richtung drehen und auseinanderziehen, sodass die Pellets in die Schüssel mit der weichen Nahrung fallen. Die Kapsel sollte vorsichtig angetippt werden, damit alle Pellets herausfallen.
3.Schritt 2 wiederholen, wenn die Dosis mehr als eine Kapsel erfordert.
4.Die Pellets vorsichtig mit einem Löffel in die Nahrung mit weicher Konsistenz einrühren.
5.Die gesamte Dosis unmittelbar nach dem Einrühren einnehmen. Das Gemisch nicht zum späteren Verzehr aufbewahren.
6.Nach Einnahme der Dosis ein Glas Wasser trinken.
7.Alle leeren Kapselhüllen entsorgen.
Einnahme in einer Flüssigkeit (erfordert eine Applikationsspritze für Zubereitungen zum Einnehmen)
Wird das Arzneimittel in einer Flüssigkeit verabreicht, so ist die Verwendung einer Applikationsspritze für Zubereitungen zum Einnehmen erforderlich. Nicht mit Hilfe einer Flasche oder Schnabeltasse geben, da die Pellets nicht durch die Öffnung passen. Die Pellets lösen sich nicht in Flüssigkeit auf.
Beim Öffnen der Kapseln zur Verteilung des Inhalts in einer Flüssigkeit sollte der Patient bzw. die Betreuungsperson angewiesen werden, wie folgt vorzugehen:
1.Die Kapsel an beiden Enden halten, in entgegengesetzte Richtungen drehen und auseinanderziehen, sodass die Pellets in ein kleines Mischgefäss fallen. Die Kapsel sollte vorsichtig angetippt werden, damit alle Pellets herausfallen.
2.Schritt 1 wiederholen, wenn die Dosis mehr als eine Kapsel erfordert.
3.1 Teelöffel (5 ml) einer altersgerechten Flüssigkeit (z.B. Muttermilch, Säuglingsnahrung oder Wasser) zugeben. Die Pellets ungefähr 5 Minuten in der Flüssigkeit belassen, damit sie sich vollsaugen.
4.Nach 5 Minuten die Spitze der Applikationsspritze vollständig in das Mischgefäss eintauchen. Kolben der Spritze langsam herausziehen, um die Mischung aus Flüssigkeit und Pellets in die Spritze aufzuziehen. Kolben anschliessend vorsichtig wieder herunterdrücken, um die Mischung aus Flüssigkeit und Pellets wieder in das Mischgefäss zu geben. Diesen Vorgang 2- bis 3-mal wiederholen, um ein vollständiges Vermischen der Pellets mit der Flüssigkeit sicherzustellen (die Pellets werden sich nicht auflösen).
5.Den gesamten Inhalt durch Herausziehen des Kolbens am Ende der Spritze in die Applikationsspritze aufziehen.
6.Die Spitze der Spritze vorne im Mund des Patienten zwischen Zunge und Wangeninnenseite platzieren und dann vorsichtig den Kolben herunterdrücken, um die Mischung aus Flüssigkeit und Pellets zwischen die Zunge und die Wangeninnenseite des Patienten zu spritzen. Die Mischung aus Flüssigkeit und Pellets nicht in den hinteren Rachenraum des Patienten spritzen, da dies zu Würgen oder Verschlucken führen könnte.
7.Wenn ein Rest der Mischung aus Flüssigkeit und Pellets in dem Mischgefäss verbleibt, wiederholen Sie die Schritte 5 und 6, bis die gesamte Dosis gegeben wurde. Das Gemisch nicht zur späteren Verwendung aufbewahren.
8.Nach Einnahme der Dosis Muttermilch, Säuglingsnahrung oder eine andere altersgerechte Flüssigkeit geben.
9.Alle leeren Kapselhüllen entsorgen.
KontraindikationenVorausgegangene oder aktive Zeichen der Leberdekompensation (z.B. Varizenblutung, Aszites, hepatische Enzephalopathie; siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Überempfindlichkeit gegen den Wirkstoff oder gegen einen der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenEnterohepatischer Kreislauf
Der Wirkmechanismus von Odevixibat setzt voraus, dass der enterohepatische Kreislauf von Gallensäuren und der Gallensalztransport in die Gallenkanälchen erhalten bleibt. Zustände, Medikamente oder chirurgische Eingriffe, die entweder die gastrointestinale Motilität oder den enterohepatischen Kreislauf der Gallensäuren beeinträchtigen, einschliesslich des Transports der Gallensalze in die Gallenkanälchen, können die Wirksamkeit von Bylvay reduzieren. Aus diesem Grund werden z.B. Patienten mit PFIC2, die ein vollständiges Fehlen oder eine mangelnde Funktion des Proteins der Gallensalzexportpumpe (BSEP) aufweisen (d.h. Patienten mit dem BSEP3-Subtyp von PFIC2), nicht auf Odevixibat ansprechen.
Es gibt nur begrenzte klinischen Daten zu Bylvay bei anderen PFIC-Subtypen als 1 und 2. Bei Patienten mit PFIC5 wurde die Wirksamkeit von Bylvay nicht untersucht.
Hepatotoxizität
Die Behandlung mit Bylvay ist mit einem Potenzial für arzneimittelinduzierte Leberschäden (DILI) verbunden. In den PFIC- und ALGS-Studien traten unter der Behandlung erhöhte Leberwerte oder eine Verschlechterung der Leberwerte auf. Von den sechs Patienten, bei denen mögliche DILI auftrat, erhielten zwei eine Lebertransplantation. Es wird empfohlen, vor Beginn der Behandlung mit Bylvay die Leberfunktionswerte zu überprüfen und eine persönliche Baseline zu etablieren; die Überwachung sollte entsprechend der klinischen Standardpraxis erfolgen. Bei Patienten mit erhöhten Leberwerten ist eine häufigere Überwachung in Betracht zu ziehen. Wenn abnorme Leberwerte oder Anzeichen einer klinischen Hepatitis ohne andere Ursachen auftreten, ist eine Dosisreduktion oder eine Unterbrechung der Behandlung in Betracht zu ziehen. Sobald die Leberfunktionsparameter wieder die Ausgangswerte erreichen, kann eine erneute Gabe von Bylvay in der empfohlenen Dosierung in Betracht gezogen werden.
Brechen Sie die Behandlung mit Bylvay dauerhaft ab, wenn bei einem Patienten Folgendes auftritt:
anhaltende oder wiederkehrende abnorme Leberwerte, oder
bei erneuter Gabe Anzeichen und Symptome, die mit einer klinischen Hepatitis vereinbar sind, oder
eine hepatische Dekompensation.
Die Sicherheit und Wirksamkeit von Bylvay bei Patienten mit dekompensierter Zirrhose wurde nicht nachgewiesen. Patienten mit kompensierter Zirrhose oder portaler Hypertonie sind häufiger zu überwachen und die Behandlung mit Bylvay ist abzubrechen, wenn eine Leberdekompensation auftritt. IBAT-Inhibitoren, einschliesslich Bylvay, sind bei Patienten mit vorausgegangenen oder aktiven Zeichen der Leberdekompensation kontraindiziert.
Diarrhö
Diarrhö wurde als häufige Nebenwirkung bei der Einnahme von Bylvay gemeldet. Diarrhö kann zu Dehydratation führen. Die Patienten sollten regelmässig überwacht werden, um eine ausreichende Flüssigkeitszufuhr während Diarrhö-Episoden sicherzustellen (siehe "Unerwünschte Wirkungen" ). Bei andauerndem Durchfall kann eine Behandlungsunterbrechung oder ein -abbruch erforderlich sein.
Mangel an fettlöslichen Vitaminen
In klinischen Studien wurden bei einigen Patienten, die Bylvay erhielten, verminderte Spiegel fettlöslicher Vitamine beobachtet. Vor Beginn der Einnahme von Bylvay wird bei allen Patienten die Beurteilung der Spiegel fettlöslicher Vitamine (FSV) (Vitamin A, D, E) und der International Normalised Ratio (INR) empfohlen; die Überwachung sollte entsprechend der klinischen Standardpraxis erfolgen. Wenn ein FSV-Mangel diagnostiziert wird, sollte eine Substitutionstherapie verordnet werden. Wenn Komplikationen aufgrund eines FSV-Mangels auftreten (z.B. Blutungen, Frakturen etc.), sollte eine Unterbrechung der Behandlung mit Bylvay in Betracht gezogen und eine Neubewertung vorgenommen werden, um eine ausreichende Supplementierung mit FSV sicherzustellen. Eine Wiederaufnahme der Behandlung mit Bylvay kann in Betracht gezogen werden, sobald der Patient klinisch stabil ist. Wenn der FSV-Mangel trotz ausreichender FSV-Supplementierung anhält oder sich verschlimmert, sollte ein dauerhafter Abbruch der Behandlung mit Bylvay in Betracht gezogen werden.
Lipophile Arzneimittel
Die Absorption lipophiler Arzneimittel kann bei gleichzeitiger Einnahme von Bylvay beeinträchtigt sein (siehe Rubrik "Interaktionen" ).
InteraktionenEinfluss anderer Substanzen auf die Pharmakokinetik von Odevixibat
Transporter-vermittelte Interaktionen
Odevixibat ist ein Substrat für den Effluxtransporter P-Glycoprotein (P-gp) aber nicht für BCRP. Bei gesunden erwachsenen Studienteilnehmern erhöhte die gleichzeitige Anwendung des starken P-gp-Inhibitors Itraconazol die Plasmaexposition einer Einzeldosis Odevixibat 7200 µg um etwa 50–60 %. Dieser Anstieg wird nicht als klinisch relevant angesehen. Es wurden keine weiteren potenziell relevanten Transporter-vermittelten Interaktionen in vitro festgestellt.
Einfluss von Odevixibat auf die Pharmakokinetik anderer Substanzen
Cytochrom-P450-vermittelte Interaktionen
In in-vitro-Studien hemmte Odevixibat die CYP-Enzyme 1A2, 2B6, 2C8, 2C9, 2C19 oder 2D6 bei klinisch relevanten Konzentrationen nicht; allerdings erwies es sich als CYP3A4/5-Inhibitor (siehe "Pharmakokinetik" ).
Bei gesunden erwachsenen Studienteilnehmern verringerte die gleichzeitige Anwendung von Odevixibat die Fläche unter der Kurve (AUC) von oralem Midazolam (einem CYP3A4-Substrat) um 30 % und die 1-OH-Midazolam-Exposition um weniger als 20 %, was nicht als klinisch relevant angesehen wird.
Es wurden keine Studien zur Erfassung von Interaktionen mit UDCA und Rifampicin durchgeführt.
In einer Studie zur Erfassung von Interaktionen mit einem Ethinylestradiol (EE) (0,03 mg) und Levonorgestrel (LVN) (0,15 mg) enthaltenden lipophilen kombinierten oralen Kontrazeptivum an erwachsenen gesunden Frauen hatte die gleichzeitige Anwendung von Odevixibat keine Auswirkungen auf die AUC von LVN und verminderte die AUC von EE um 17 %, was als klinisch nicht relevant erachtet wird. Es wurden keine Studien zur Erfassung von Interaktionen mit anderen lipophilen Arzneimitteln durchgeführt, weshalb eine Wirkung auf die Absorption anderer fettlöslicher Arzneimittel nicht ausgeschlossen werden kann.
In klinischen Studien wurden bei einigen Patienten, die Odevixibat erhielten, reduzierte Spiegel fettlöslicher Vitamine beobachtet. Die Spiegel fettlöslicher Vitamine sollten überwacht werden.
In-vitro-Studien
Odevixibat hemmte nicht die Transporter P-gp, BCRP, organische Anionen-Transport-Polypeptide 1B1 und 1B3 (OATP1B1 und OATP1B3); organische Anionentransporter (OAT)1, OAT3; organische Kationentransporter 2 (OCT2), Multidrug- und Toxin-Extrusions-Transporter 1 und 2K (MATE1 und MATE2K).
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Frauen im gebärfähigen Alter sollten eine zuverlässige Verhütungsmethode anwenden, wenn sie mit Bylvay behandelt werden.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Bylvay bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe "Präklinische Daten" ). Die Anwendung von Bylvay während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Stillzeit
Es ist nicht bekannt, ob Odevixibat oder seine Metaboliten in die Muttermilch übergehen. Es gibt nur ungenügende Informationen darüber, ob Odevixibat beim Tier in die Milch übergeht (siehe "Präklinische Daten" ).
Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Bylvay verzichtet werden soll / die Behandlung mit Bylvay zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Mutter zu berücksichtigen.
Fertilität
Es liegen keine Daten zur Fertilität beim Menschen vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte Auswirkungen auf die Fertilität oder Reproduktion (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenBylvay hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die am häufigsten gemeldete Nebenwirkung im klinischen Entwicklungsprogramm war Diarrhö bei 31 % der Patienten mit PFIC und 37 % der Patienten mit ALGS. Sonstige gemeldete Nebenwirkungen waren Erbrechen, Magenschmerzen, Erhöhungen der Leberwerte, Hepatomegalie und Abnahme der Vitamin-D und Vitamin-E Spiegel.
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen werden nach Systemorganklassen eingeteilt und wie folgt klassifiziert: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1000, < 1/100), selten (≥1/10 000, < 1/1000), sehr selten (< 1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
In der Tabelle sind die Nebenwirkungen aufgelistet, die in klinischen Studien an Patienten mit PFIC im Alter von 4 Monaten bis 25 Jahren (Median: 3 Jahre 7 Monate) und in klinischen Studien an Patienten mit ALGS im Alter von 1 bis zu 16 Jahren (Median: 5,7 Jahre) identifiziert wurden.
Tabelle 3: Häufigkeit der Nebenwirkungen bei PFIC- und ALGS-Patienten
MedDRA-Systemorgankla PFIC ALGS
sse Häufigkeit
Erkrankungen des
Gastrointestinaltrakt
s
-sehr häufig Diarrhöa (31 %), Erbrechen (17 Diarrhöa (37 %), Abdominalschmerzb (17 %)
%), Abdominalschmerzb (11 %)
-häufig - Erbrechen
Leber- und Gallenerkr
ankungen
-sehr häufig Bilirubin im Blut erhöht (25 -
%), ALT erhöht (14 %)
-häufig Hepatomegalie, AST erhöht Bilirubin im Blut erhöht, ALT erhöht,
AST erhöht, GGT erhöht, Hepatomegalie
Stoffwechsel- und
Ernährungsstörungen
-sehr häufig Vitamin-D-Mangel (11 %) Vitamin-D-Mangel (14 %)
-häufig Vitamin-E-Mangel Vitamin-E-Mangel
a Basierend auf der kombinierten Häufigkeit von Diarrhö, hämorrhagische Diarrhö und weicher Stuhl.
b Beinhaltet Schmerzen Oberbauch und Schmerzen Unterbauch
ALT = Alanin-Aminotransferase
AST = Aspartat-Aminotransferase
GGT = Gamma-Glutamyltransferase
Beschreibung ausgewählter Nebenwirkungen
Gastrointestinale Nebenwirkungen
PFIC
Die am häufigsten berichtete gastrointestinale Nebenwirkung bei etwa 31 % der mit Bylvay behandelten Patienten in den klinischen Studien war Diarrhö. Die meisten Ereignisse von Diarrhö waren von kurzer Dauer (≤5 Tage), von leichter bis mittlerer Intensität (99 %) und nicht schwerwiegend. Dosisreduktion (3 %), Behandlungsunterbrechung (7 %) und -abbruch (3 %) aufgrund von Diarrhö wurden bei wenigen Patienten gemeldet, die aufgrund von Diarrhö eine intravenöse oder orale Hydratation benötigten (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Andere häufig berichtete gastrointestinale Nebenwirkungen waren Erbrechen (17 %) und Abdominalschmerzen (11 %) (einschliesslich Schmerzen im Ober- und Unterbauch), die alle nicht schwerwiegend, leicht bis mittelschwer waren und im Allgemeinen keine Dosisanpassung erforderten.
ALGS
Die am häufigsten gemeldete Nebenwirkung bei 37 % der mit Bylvay behandelten Patienten war Diarrhö. Alle Vorkommnisse waren leicht bis mittelschwer und nicht schwerwiegend. Nur wenige Patienten (4 %) mussten die Behandlung unterbrechen und eine Rehydrierung aufgrund von Diarrhö durchführen (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Andere gemeldete gastrointestinale Nebenwirkungen waren Abdominalschmerzen (17 %) und Erbrechen (6 %), leicht bis mittelschwer und von begrenzter Dauer.
Leber- und Gallenerkrankungen
PFIC
Die häufigsten hepatischen Nebenwirkungen waren erhöhte Bilirubin- (25 %), Alaninaminotransferase (ALT) (14 %)- und Aspartataminotransferase (AST)-Werte (9 %) im Blut. Die meisten davon waren leicht bis mittelschwer; bei fünf Patienten (4 %) wurde von stark erhöhten Leberwerten berichtet. Bei mit Bylvay behandelten PFIC-Patienten wurden Behandlungsunterbrechungen aufgrund von erhöhten Leberwerten berichtet. Die Behandlung mit Bylvay ist mit einem Potenzial für arzneimittelinduzierte Leberschäden (DILI) verbunden. Jedoch waren die meisten Abweichungen der ALT, AST und Bilirubinwerte auch auf die Grunderkrankung sowie auf zeitweise auftretende virale oder infektiöse Begleiterkrankungen zurückzuführen, die bei der Altersgruppe der Patienten häufig sind. Daher wird die Überwachung der Leberwerte empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
ALGS
Die häufigsten hepatischen Nebenwirkungen waren erhöhte ALT (6 %) -, AST-, GGT-, und Bilirubinwerte (alle 4 %) im Blut. Die meisten dieser Abweichungen waren leicht bis mässig und nicht schwerwiegend. Die Behandlung mit Bylvay ist mit einem Potenzial für arzneimittelinduzierte Leberschäden (DILI) verbunden. Die meisten erhöhten Leberenzym- und Bilirubinwerte wurden aufgrund der ALGS-bedingten zugrundeliegenden Pathophysiologie der Leber beobachtet. Daher wird die Überwachung der Leberwerte empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Stoffwechsel- und Ernährungsstörungen
PFIC und ALGS
Aufgrund der verminderten Ausscheidung von Gallensäuren in den Darm und des Risikos einer Malabsorption besteht bei pädiatrischen Patienten mit PFIC und ALGS mit chronischer Cholestase auch bei Supplementierung das Risiko eines Mangels an fettlöslichen Vitaminen (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Während der Langzeitbehandlung mit Bylvay wurden verminderte Vitaminspiegel beobachtet; die Mehrheit dieser Patienten sprach auf eine angemessene Vitaminergänzung an. Insgesamt wurde bei wenigen Patienten ein Mangel an fettlöslichen Vitaminen festgestellt, der sich nicht durch eine Nahrungsergänzung beheben liess (8 % bei ALGS und 3 % bei PFIC Patienten). Diese Ereignisse waren von geringer Intensität und führten nicht zu einer Unterbrechung der Behandlung oder einem Absetzen von Bylvay.
Erfahrungen seit Markteinführung
Die unerwünschten Wirkungen, welche seit Markteinführung berichtet wurden, waren konsistent mit den unerwünschten Wirkungen, welche in klinischen Studien beobachtet worden waren. Die Datenlage reicht nicht aus, um die Inzidenz in der PFIC- und der ALGS-Population zu schätzen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEine Überdosierung kann zu Symptomen führen, die durch ein übermässig starkes Auftreten der bekannten pharmakodynamischen Wirkungen des Arzneimittels bedingt sind. Diese sind vor allem Diarrhö und gastrointestinale Wirkungen.
Die maximale Dosis, die gesunden Studienteilnehmern in klinischen Studien gegeben wurde, war Bylvay 10 000 µg als Einzeldosis ohne unerwünschte Folgen.
Im Falle einer Überdosierung ist der Patient symptomatisch zu behandeln, und unterstützende Massnahmen sind nach Bedarf einzuleiten.
Eigenschaften/WirkungenATC-Code
A05AX05
Wirkungsmechanismus
Odevixibat ist ein reversibler, selektiver Inhibitor des ilealen Gallensäuretransporters (IBAT).
Pharmakodynamik
Odevixibat wirkt lokal im distalen Ileum. Es verringert die Wiederaufnahme der Gallensäuren, erhöht die Clearance der Gallensäuren über den Dickdarm und reduziert so die Gallensäurekonzentration im Serum. Das Ausmass der Reduktion der Gallensäuren im Serum korreliert nicht mit der systemischen Pharmakokinetik.
Klinische Wirksamkeit
PFIC
Die Wirksamkeit von Bylvay bei Patienten mit PFIC wurde in einer Phase-III-Studie (Studie A4250-005) untersucht. Bei dieser PFIC-Studie 1 handelte es sich um eine 24-wöchige, randomisierte, doppelblinde, placebokontrollierte Studie, an der 62 Patienten mit bestätigter PFIC-Typ-1- oder PFIC-Typ-2-Diagnose und signifikantem Pruritus in der Vorgeschichte sowie bestehendem Pruritus bei Studienbeginn teilnahmen. Die Patienten wurden 1:1:1 randomisiert und erhielten Placebo, Bylvay 40 oder 120 µg/kg/Tag und wurden nach PFIC-Typ (1 oder 2) und Alter (6 Monate bis 5 Jahre, 6 bis 12 Jahre und 13 bis ≤18 Jahre) stratifiziert. Patienten mit pathologischen Variationen des ABCB11-Gens, die das vollständige Fehlen des BSEP-Proteins prognostizieren, und Patienten mit ALT-Konzentrationen > 10 × ULN oder Bilirubin-Konzentrationen > 10 × ULN wurden ausgeschlossen. Bei 13 % der Patienten war zuvor eine biliäre Diversionsoperation durchgeführt worden. Der primäre Endpunkt der Studie PFIC 1 war der Anteil der positiven Pruritus-Bewertungen auf Patientenebene über den 24-wöchigen Behandlungszeitraum basierend auf einem von einem Beobachter festgestellten Ergebnis ( "observer-reported outcome" , ObsRO). Mit ObsRO wurde der Schweregrad des Kratzens auf einer 5-Punkte-Skala (0–4) beurteilt, wobei die Werte von 0 (kein Kratzen) bis 4 (schwerstmögliches Kratzen) reichten. Eine positive Pruritus-Bewertung war ein Wert von ≤1 oder eine Verbesserung um mindestens 1 Punkt gegenüber dem Ausgangswert. Pruritus-Bewertungen wurden morgens und abends durchgeführt. Ein zusätzlicher primärer Endpunkt war der Anteil an Patienten, bei denen nach 24 Wochen der Behandlung der Gallensäurespiegel im Serum (nüchtern) um mindestens 70 % gesenkt wurde oder einen Wert von ≤70 µmol/l erreichte.
Das Medianalter (Spanne) der PFIC-Patienten in Studie 1 betrug 3,2 (0,5 bis 15,9) Jahre; 50 % der Patienten waren männlich und 84 % weiss. 27 % der Patienten wiesen einen PFIC-Typ 1 auf und 73 % hatten den PFIC-Typ 2. Zu Studienbeginn (Baseline) wurden 81 % der Patienten mit UDCA, 66 % mit Rifampicin und 89 % mit UDCA und/oder Rifampicin behandelt. Die Baseline der Leberbeeinträchtigung nach Child-Pugh-Klassifikation war bei 66 % der Patienten leicht und bei 34 % der Patienten mittelschwer. Der mittlere Ausgangswert (Standardabweichung, SD) der eGFR betrug 164 (30,6) ml/min/1,73 m². Die mittleren Ausgangswerte (SD) der ALT-, AST- und Bilirubin-Werte betrugen 99 (116,8) E/l, 101 (69,8) U/l bzw. 3,2 (3,57) mg/dl. Die mittlere Baseline (SD) des Pruritus-Scores (Spanne: 0−4) und der Gallensäurespiegel im Serum waren bei den mit Odevixibat behandelten Patienten (2,9 [0,089] bzw. 252,1 [103,0] µmol/l) ähnlich wie bei den mit Placebo behandelten Patienten (3,0 [0,143] bzw. 247,5 [101,1] µmol/l).
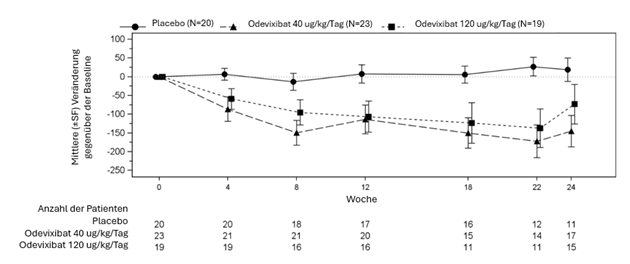
Tabelle 4 zeigt die Ergebnisse des Vergleichs der wichtigsten Wirksamkeitsdaten in PFIC-Studie 1 zwischen Odevixibat und Placebo. Die Daten bezüglich des Pruritus und des Gallensäurespiegels im Serum sind über die 24-wöchige Behandlungsdauer grafisch in Abbildung 1 (Anteil der Pruritus Bewertungen mit Ergebnissen von 0 oder 1) und in Abbildung 2 (mittlere Veränderung der Gallensäurekonzentration im Serum verglichen zur Baseline) dargestellt.
Tabelle 4: Vergleich der wichtigsten Wirksamkeitsergebnisse für Odevixibat und Placebo während des 24-wöchigen Behandlungszeitraums (PFIC-Studie 1)
Wirksamkeitsendpunkt Placebo (N = 20) Odevixibat
40 µg/kg/Tag (N = 23) 120 µg/kg/Tag (N =
19)
Anteil der positiven Pruritus-Bewe
rtungen während des Behandlungszei
traums
Anteil 28,74 58,31 47,69
Relativer Unterschied (SF) im 28,23 (9,18) (9,83; 21,71 (9,89) (1,87;
Vergleich zu Placebo (95%-KI)a 46,64) 41,54)
Einseitiger p-Wertc 0,0019 0,0163
Anteil der Patienten mit
reduzierten Gallensäurespiegeln
im Serum am Ende der Behandlung
(Respondersb)
N (%) (95%-KI) 0 (0,00; 16,84) 10 (43,5) (23,19; 4 (21,1) (6,05;
65,51) 45,57)
Relativer Unterschied im 0,44 (0,22; 0,66) 0,21 (0,02; 0,46)
Vergleich zu Placebo (95%-KI)
Einseitiger p-Wertc 0,0015 0,0174
a Basiert auf dem Kleinstquadrat-Mittelwert (least squares means) aus einer Kovarianzanalyse, wobei die Baseline-Pruritus-Werte (Tag und Nacht) als Kovariaten und die Behandlungsgruppe und Stratifikationsfaktoren (PFIC-Typ und Altersgruppe) als fixe Effekte eingestuft werden.
b Responders wurden definiert als eine mindestens 70-prozentige Verringerung der Serumgallensäurekonzentration gegenüber dem Ausgangswert oder das Erreichen eines Wertes ≤ 70 µmol/L.
c Basiert auf dem nach PFIC-Typ stratifizierten Cochran-Mantel-Haenszel-Test. Die p-Werte für die Dosisgruppen wurden hinsichtlich Multiplizität angepasst.
Abbildung 1: Anteil der Pruritus-Bewertungen auf Patientenebene mit Pruritus Scores von 0 oder 1 über den 24-wöchigen Behandlungszeitraum in 4-Wochen Intervallen (ObsRO (observer-reported outcome) Instrument)
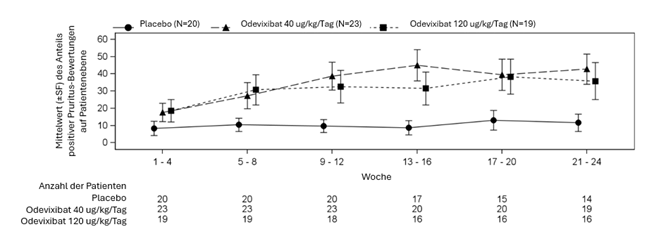
Abbildung 2: Mittlere (±SF) Veränderung der Gallensäurekonzentration im Serum (μmol/l) gegenüber der Baseline im Laufe der Zeit
Patienten, welche die PFIC-Studie 1 abschlossen, konnten an der PFIC-Studie 2 (Studie A4250-008) teilnehmen, einer offenen Verlängerungsstudie, in der alle Patienten Odevixibat 120 µg/kg/Tag für weitere 72 Wochen erhielten. Im Durchschnitt blieben die in PFIC-Studie 1 bei Patienten unter Behandlung mit Odevixibat beobachteten Verbesserungen hinsichtlich Pruritus und Serum-Gallensäuren 72 Wochen erhalten.
ALGS
Die Wirksamkeit von Bylvay bei Patienten mit ALGS wurde in einer Phase-III-Studie (Studie A4250-012) bewertet. Bei ALGS-Studie 1 handelte es sich um eine 24-wöchige, randomisierte, doppelblinde, placebokontrollierte Studie, an der 52 Patienten mit bestätigter ALGS-Diagnose und signifikantem Pruritus in der Vorgeschichte sowie bestehendem Pruritus bei Studienbeginn teilnahmen. Die Patienten wurden per Randomisierung im Verhältnis 2:1 einer Behandlung mit 120 µg/kg/Tag Odevixibat oder Placebo zugeteilt, stratifiziert nach Alter bei Randomisierung (< 10 Jahre und ≥10 bis < 18 Jahre). Patienten mit ALT > 10 × ULN oder Gesamtbilirubin > 15 × ULN beim Screening wurden von der ALGS-Studie 1 ausgeschlossen.
Der primäre Endpunkt in ALGS-Studie 1 war die Veränderung des Pruritus-Scores zwischen der Baseline und Monat 6 (Wochen 21 bis 24). Pruritus wurde einmal morgens und einmal abends durch einen Beobachter bewertet ( "observer-reported outcome" , ObsRO). Mit ObsRO wurde der Schweregrad des Kratzens auf einer 5-Punkte-Skala (0–4) beurteilt, wobei die Werte von 0 (kein Kratzen) bis 4 (schwerstmögliches Kratzen) reichten.
Als wichtigster sekundärer Endpunkt diente die Veränderung des Gallensäurespiegels im Serum zwischen der Baseline und dem Durchschnitt in den Wochen 20 und 24.
Das Medianalter (Spanne) der Patienten in ALGS-Studie 1 betrug 5,45 (0,5 bis 15,5) Jahre; 51,9 % der Patienten waren männlich und 82,7 % weiss. 92,3 % der Patienten wiesen die JAG1-Mutation und 7,7 % die NOTCH2-Mutation auf. Bei Baseline erhielten 98,1 % der Patienten antipruritische Begleitmedikationen, darunter UDCA (88,5 %). Insgesamt litten 51 (98,1 %) der 52 Patienten an einer mittelschweren Leberfunktionsstörung, und bei 1 Patienten (1,9 %) (Placebogruppe) lag eine schwere Leberfunktionsstörung vor (auf Basis der Child-Pugh-Klassifikation). Der mittlere Ausgangswert (SD) der eGFR betrug 158,65 (51,437) ml/min/1,73 m². Die mittleren Ausgangswerte (SD) von ALT, AST und Gesamtbilirubin betrugen 173,7 (84,48) U/l, 167,0 (83,22) U/l bzw. 55,14 (47,911) µmol/l. Der mittlere Baseline-Wert (SD) des Pruritus-Scores (Spanne: 0–4) und der Gallensäurespiegel im Serum waren bei den mit Odevixibat behandelten Patienten (2,80 [0,520] bzw. 237,4 [114,88] µmol/l) ähnlich wie bei den mit Placebo behandelten Patienten (3,01 [0,636] bzw. 246,1 [120,53] µmol/l).
Tabelle 5 zeigt die Veränderung des mittleren Pruritus-Scores auf Grundlage von ObsRO-Bewertungen zwischen der Baseline und Monat 6 (Wochen 21 bis 24) und die Veränderung des Gallensäurespiegels im Serum zwischen der Baseline und dem Durchschnitt in den Wochen 20 und 24.
Tabelle 5: Vergleich der wichtigsten Wirksamkeitsergebnisse für Odevixibat und Placebo während des 24-wöchigen Behandlungszeitraums (ALGS-Studie 1)
Placebo(N = 17) Odevixibat120
µg/kg/Tag(N = 35)
Veränderung des mittleren Pruritus-Scores zwischen
Baseline und Monat 6 (Wochen 21 bis 24) der Behandlung
LS-Mittelwert (SF)a -0,80 (0,233) -1,69 (0,174)
LS-Mittelwert-Differenz vs. Placebo -0,88 (-1,44;
(95%-KI)aEinseitiger p-Werta -0,33)0,0012
Veränderung des Gallensäurespiegels im Serum (µmol/l)
zwischen Baseline und dem Durchschnitt in den Wochen 20
und 24 der Behandlung
LS-Mittelwert (SF)a 22,39 (28,463) -90,35 (21,336)
LS-Mittelwert-Differenz vs. Placebo -112,74 (-178,78;
(95%-KI)aEinseitiger p-Werta -46,69)0,0006
LS-Mittelwert = Kleinstquadrat-Mittelwert
a Die Analysen basieren auf einem Mischmodell für Wiederholungsmessungen (MMRM) mit Pruritus-Score bei Baseline oder Gallensäurekonzentration im Serum bei Baseline (wie für den Endpunkt zutreffend) als Kovariante und Stratifikation nach Alter bei Baseline (< 10, ≥10 Jahre), direktes Bilirubin bei Baseline (nur Pruritus-Score), Behandlungsgruppe, Zeit (Monate/Besuche) und Interaktion zwischen Behandlung und Zeit als fixe Effekte.
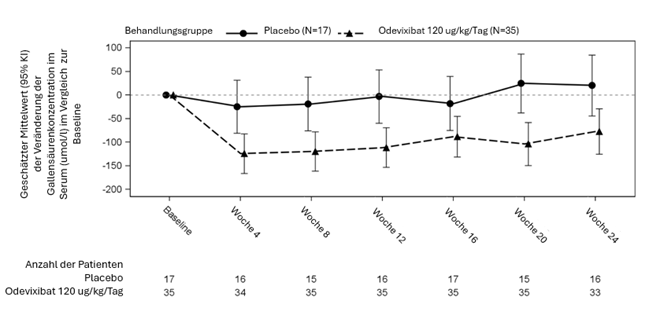
In den Abbildungen 3 und 4 sind die mittleren Veränderungen (SF) des durchschnittlichen Pruritus-Scores der Patienten bzw. der Gallensäurespiegel im Serum der Patienten in jeder Behandlungsgruppe für jeden Monat gegenüber dem Ausgangswert dargestellt.
Abbildung 3: Mittlere (± SF) Veränderung des Schweregrads des Pruritus gegenüber der Baseline im Laufe der Zeit (ALGS Studie 1)
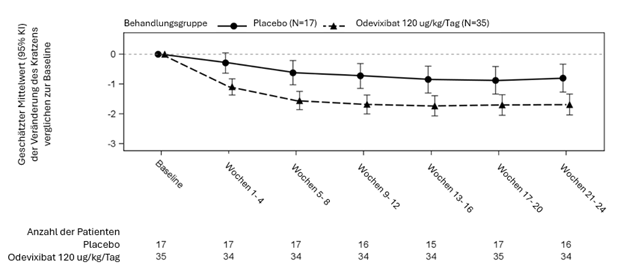
Abbildung 4: Mittlere (± SF) Veränderung der Gallensäurekonzentration im Serum (µmol/l) gegenüber der Baseline im Laufe der Zeit (ALGS Studie 1)
Patienten, welche die ALGS-Studie 1 abgeschlossen hatten, konnten an der ALGS-Studie 2 (Studie A4250-015) teilnehmen, einer 72-wöchigen offenen Verlängerungsstudie. Im Durchschnitt blieben die in ALGS-Studie 1 bei Patienten unter Behandlung mit Odevixibat beobachteten Verbesserungen hinsichtlich Pruritus und Serum-Gallensäuren 72 Wochen erhalten.
PharmakokinetikAbsorption
Odevixibat wird nach oraler Gabe minimal absorbiert; es liegen keine Daten zur absoluten Bioverfügbarkeit beim Menschen vor, und die geschätzte relative Bioverfügbarkeit beträgt < 1,5 %. Die maximale Plasmakonzentration von Odevixibat (Cmax) wird innerhalb von 1 bis 5 Stunden erreicht. Die beobachteten Expositionen bei pädiatrischen Patienten (Alter zwischen 1,1 und 16,0 Jahren; Körpergewicht zwischen 5,6 und 55,2 kg) sind auf Talspiegel begrenzt. Für die Dosis von 120 µg/kg/Tag lagen die Talspiegel bei 88 % der Proben von Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) und bei 43 % der Proben von Patienten mit mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse B) unterhalb der Nachweisgrenze. Die maximalen Talkonzentrationen, die bei Child-Pugh-Klasse A und B beobachtet wurden, betrugen 0,455 bzw. 3,38 ng/ml. Simulierte Cmax-Werte in einer pädiatrischen PFIC-Patientenpopulation für die Dosen 40 µg/kg/Tag und 120 µg/kg/Tag betrugen 0,211 ng/ml bzw. 0,623 ng/ml; die AUC-Werte betrugen 2,26 ng × h/ml bzw. 5,99 ng × h/ml. Die simulierten Cmax- und AUC-Werte für die Dosis von 120 µg/kg/Tag fielen in einer pädiatrischen ALGS-Population ähnlich aus wie bei PFIC. Es besteht nur eine minimale Akkumulation von Odevixibat nach einmal täglicher Einnahme.
Nahrungseinfluss
Die systemische Exposition von Odevixibat lässt keine Wirksamkeitsprognose zu. Daher wird keine Dosisanpassung im Hinblick auf die Auswirkungen der Einnahme von Nahrung als notwendig erachtet. Die gleichzeitige Gabe einer fettreichen Mahlzeit (800–1000 Kalorien mit ca. 50 % des gesamten Kaloriengehalts der Mahlzeit aus Fett) führte im Vergleich zur Anwendung unter Nüchternbedingungen zu einer Reduzierung der Cmax um ca. 72 % und der AUC0–24 um ca. 62 %. Wenn Odevixibat auf Apfelmus gestreut wurde, wurden gegenüber der Gabe unter Nüchternbedingungen eine Reduzierung der Cmax und der AUC0–24 um ca. 39 % bzw. 36 % beobachtet. Da keine PK/PD-Beziehung besteht und Odevixibat bei jüngeren Kindern auf die Nahrung gestreut werden muss, kann Odevixibat mit Nahrung eingenommen werden.
Distribution
Odevixibat bindet zu über 99 % an humane Plasmaproteine. Das mittlere an das Körpergewicht angepasste apparente Verteilungsvolumen (V/F) beträgt bei pädiatrischen PFIC-Patienten für die Dosierungsschemata 40 µg/kg/Tag und 120 µg/kg/Tag 40,3 bzw. 43,7 l/kg. Bei einer typischen 70 kg schweren Person wird das V/F auf 3338 l geschätzt. Das mittlere Verteilungsvolumen (V/F) wird bei ALGS-Patienten auf 1160 l geschätzt. Der geometrische Mittelwert des körpergewichtsbereinigten V/F für ALGS beträgt 57,9 l/kg.
Metabolismus
Odevixibat wird bei Menschen minimal metabolisiert.
Elimination
Nach Gabe einer oralen Einzeldosis von 3000 μg radioaktiv markiertem Odevixibat an gesunde Erwachsene betrug die durchschnittliche prozentuale Rückgewinnung der gegebenen Dosis 82,9 % im Stuhl; weniger als 0,002 % wurden aus dem Urin wiedergewonnen. Es wurde ermittelt, dass über 97 % der Radioaktivität im Stuhl auf unverändertes Odevixibat zurückzuführen war.
Die auf das Körpergewicht normalisierte apparente Gesamt-Clearance (CL/F) beträgt bei pädiatrischen PFIC-Patienten für die Dosierungsschemata 40 µg/kg/Tag und 120 µg/kg/Tag 26,4 bzw. 23,0 l/kg. Bei einer typischen 70 kg schweren Person wird die CL/F auf 2970 l/h geschätzt, und die mittlere Halbwertszeit beläuft sich auf ungefähr 2,5 Stunden. Die normalisierte apparente Gesamt-Clearance (CL/F) wird bei ALGS-Patienten auf 212 l/h geschätzt, und die mittlere Halbwertszeit beträgt etwa 4,75 Stunden. Der geometrische Mittelwert der körpergewichtsbereinigten CL/F für ALGS beträgt 10,5 l/h/kg.
Linearität/Nicht-Linearität
Die Cmax und AUC0–t steigen mit höheren Dosen dosisproportional an; jedoch kann die Dosis-Proportionalität aufgrund der hohen interindividuellen Variabilität von etwa 40 % nicht exakt geschätzt werden.
Pharmakokinetische/pharmakodynamische Zusammenhänge
Entsprechend dem Wirkungsmechanismus und dem Wirkort von Odevixibat im Gastrointestinaltrakt wird kein Zusammenhang zwischen systemischer Exposition und klinischen Wirkungen beobachtet. Auch für den untersuchten Dosisbereich von 10–200 µg/kg/Tag und die PD-Parameter C4 und FGF19 konnte kein Zusammenhang zwischen Dosis und Wirkung nachgewiesen werden.
Kinetik spezieller Patientengruppen
Basierend auf Alter, Geschlecht oder ethnischer Herkunft wurden keine klinisch signifikanten Unterschiede hinsichtlich der Pharmakokinetik von Odevixibat beobachtet.
Leberfunktionsstörung
Die Mehrzahl der Patienten mit PFIC und alle Patienten mit ALGS wiesen aufgrund dieser Erkrankung einen gewissen Grad an Leberfunktionsstörung auf. Die Verstoffwechselung von Odevixibat in der Leber spielt bei der Elimination von Odevixibat keine wesentliche Rolle. Für Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) liegen keine Daten vor.
Nierenfunktionsstörung
Es liegen keine klinischen Daten zur Anwendung von Odevixibat bei Patienten mit mittelschwerer bis schwerer Nierenfunktionsstörung oder terminaler dialysepflichtiger Niereninsuffizienz (ESRD) vor. Die Auswirkungen einer Nierenfunktionsstörung dürften aufgrund der geringen systemischen Exposition und der Tatsache, dass Odevixibat nicht über den Urin ausgeschieden wird, gering sein.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität und Kanzerogenität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Reproduktions- und Entwicklungstoxizität
Bei trächtigen Kaninchen der Rasse Weisse Neuseeländer wurde bei zwei Kaninchen, die während des Zeitraums der fetalen Organogenese Odevixibat erhielten, eine vorzeitige Entbindung / ein Abort beobachtet; die Exposition war um einen Faktor ≥2,3 höher als die erwartete klinische Exposition (basierend auf der Odevixibat-AUC0–24 im Plasma insgesamt). In allen Dosisgruppen wurde ein Rückgang des Körpergewichts und der Nahrungsaufnahme des Mutterkaninchens festgestellt (vorübergehend beim 1,1-Fachen der Exposition bei der erwarteten Dosis).
Ab dem 1,1-Fachen der humantherapeutischen Exposition (basierend auf der Odevixibat-AUC0–24 im Plasma insgesamt) wurden bei 7 Föten (1,3 % aller gegenüber Odevixibat exponierten Föten) in allen Dosisgruppen kardiovaskuläre Defekte (d.h. ventrikuläres Divertikel, kleiner Ventrikel und dilatierter Aortenbogen) festgestellt. Derartige Fehlbildungen wurden bei trächtigen Ratten, denen Odevixibat verabreicht wurde, nicht beobachtet. Aufgrund der Erkenntnisse bei Kaninchen kann eine Wirkung von Odevixibat auf die kardiovaskuläre Entwicklung nicht ausgeschlossen werden.
Odevixibat hatte in Studien bei Ratten bei dem 133-Fachen der erwarteten klinischen Exposition (basierend auf der Odevixibat-AUC0–24 im Plasma insgesamt), einschliesslich bei Jungtieren (beim 63-Fachen der zu erwartenden humantherapeutischen Exposition), keine Auswirkungen auf die Fortpflanzungsleistung, Fertilität‚ embryofetale Entwicklung oder pränatale/postnatale Entwicklung.
Es gibt nur ungenügende Informationen darüber, ob Odevixibat beim Tier in die Milch übergeht.
Das Vorhandensein von Odevixibat in der Muttermilch wurde in tierexperimentellen Studien nicht gemessen. Die Exposition wurde an den Jungtieren von laktierenden Muttertieren in der Studie zur prä- und postnatalen Entwicklungstoxizität an Ratten nachgewiesen (3,2–52,1 % der Odevixibat-Plasmakonzentration bei den laktierenden Muttertieren). Daher ist es möglich, dass Odevixibat in der Muttermilch enthalten ist.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Bei 15-30 °C lagern.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer69600 (Swissmedic)
PackungenFlasche aus hochdichtem Polyethylen (HDPE) mit kindergesichertem Originalitätsverschluss.
Hartkapseln zu 200 µg: 30 (B)
Hartkapseln zu 400 µg: 30 (B)
Hartkapseln zu 600 µg: 30 (B)
Hartkapseln zu 1200 µg: 30 (B)
ZulassungsinhaberinIPSEN Pharma Schweiz GmbH, Zug
Stand der InformationJuli 2025
|