Eigenschaften/WirkungenATC-Code
A05AX07
Wirkungsmechanismus
Seladelpar ist ein Agonist des Peroxisom-Proliferator-aktivierten Rezeptors delta (PPARδ) oder Delpar. PPARδ ist ein Kernrezeptor, der in der Leber und in anderen Geweben exprimiert wird. Die Aktivierung von PPARδ verringert die Gallensäuresynthese in der Leber durch die Fibroblasten-Wachstumsfaktor 21 (Fibroblast Growth Factor 21, FGF21)-abhängige Herunterregulierung von CYP7A1, dem wichtigsten Enzym für die Gallensäuresynthese aus Cholesterin, und durch die Verringerung der Cholesterinsynthese und -resorption.
Pruritus ist ein häufiges Symptom bei Patienten mit PBC, die Ursachen sind aber nicht vollständig geklärt. Die Behandlung mit Seladelpar hat eine Reduzierung des Pruritus gezeigt.
Pharmakodynamik
In kontrollierten klinischen Studien führte die Behandlung mit Seladelpar zu einer Reduktion von ALP, einem Biomarker der Cholestase. Eine ALP-Reduktion wurde innerhalb von 1 Woche nach Behandlungsbeginn beobachtet, nahm bis Monat 3 weiter ab und hielt bis Monat 12 an.
Kardiale Elektrophysiologie
Bei dem 20-Fachen der empfohlenen Dosis von 10 mg führte Seladelpar zu keinen klinisch signifikanten Verlängerungen des QTc-Intervalls.
Klinische Wirksamkeit
Studie CB8025-32048 – RESPONSE
Die Wirksamkeit von Lyvdelzi wurde in der 52-wöchigen, randomisierten, doppelblinden, placebokontrollierten Studie CB8025-32048 untersucht. An der Studie nahmen 193 erwachsene Patienten mit PBC teil, mit unzureichendem Ansprechen oder Unverträglichkeit gegenüber UDCA. Patienten wurden in die Studie aufgenommen, wenn ihr ALP-Spiegel ≥1,67 × ULN und ihr Gesamtbilirubin (TB)-Wert ≤2 × ULN betrug. Patienten mit anderen chronischen Lebererkrankungen, klinisch bedeutender hepatischer Dekompensation, einschliesslich portaler Hypertonie mit Komplikationen, oder Zirrhose mit Komplikationen (z.B. Modell für Lebererkrankungen im Endstadium [MELD]-Score 12 oder höher, bekannte Ösophagusvarizen oder Varizenblutungen in der Vorgeschichte, hepatorenales Syndrom in der Vorgeschichte) wurden von der Studie ausgeschlossen.
Die Patienten wurden randomisiert (2:1) und erhielten 12 Monate lang einmal täglich entweder Lyvdelzi (n=128) 10 mg oder Placebo (n=65). In der Studie erhielten 181 (94%) Patienten Lyvdelzi oder Placebo in Kombination mit UDCA, und 12 (6%) Patienten mit einer Unverträglichkeit gegenüber UDCA erhielten Lyvdelzi oder Placebo als Monotherapie.
Die beiden Behandlungsgruppen waren hinsichtlich der demographischen Merkmale und der Krankheitsmerkmale zu Studienbeginn insgesamt ausgewogen. Das mittlere Alter der Patienten betrug 57 Jahre (Spanne: 28 bis 75), 95% waren weiblich, 88% waren Weisse, 6% waren asiatischer Abstammung, 2% waren Schwarze oder Afroamerikaner und 3% waren amerikanische Indianer oder Ureinwohner Alaskas. Insgesamt 29% der Patienten, d.h. 23% im Arm mit Lyvdelzi 10 mg und 42% im Placebo-Arm, waren hispanischer/lateinamerikanischer Abstammung.
Bei Studienbeginn erfüllten 18 (14%) der mit Lyvdelzi und 9 (14%) der mit Placebo behandelten Patienten mindestens eines der folgenden Kriterien für eine Zirrhose: Fibroscan > 16,9 kPa, Biopsie oder radiologischer Befund in der Vorgeschichte mit Hinweis auf eine Zirrhose, Thrombozytenzahl < 140'000/μl mit mindestens einem zusätzlichen Laborbefund, darunter Serumalbumin < 3,5 g/dl, INR > 1,3 oder Gesamtbilirubin > 1 × ULN oder klinische Feststellung einer Zirrhose durch den Prüfarzt bzw. die Prüfärztin. Alle Zirrhose-Patienten hatten bei Studienbeginn einen Child-Pugh-Klasse A Status.
Die mittlere ALP-Konzentration zu Studienbeginn betrug 314 Einheiten pro Liter (E/l) (Spanne: 161 bis 786), entsprechend 2,7 × ULN. Die mittlere Konzentration an Gesamtbilirubin zu Studienbeginn betrug 0,8 mg/dl (Spanne: 0,3 bis 1,9) und war bei 87% der Patienten ≤ ULN. Weitere mittlere biochemische Leberwerte zu Studienbeginn betrugen: 48 E/l (Spanne: 9 bis 115) für ALT, entsprechend dem 1,2 × ULN, 40 E/l (Spanne: 16 bis 94) für AST, entsprechend dem 1,2 × ULN, und 288 E/l (Spanne: 42 bis 1088) für Gamma-Glutamyltransferase (GGT), entsprechend dem 1,7 × ULN.
Lyvdelzi zeigte eine signifikant grössere Verbesserung des biochemischen Ansprechens und der ALP-Normalisierung in Monat 12 im Vergleich zu Placebo. Die Behandlung mit Lyvdelzi führte zu einem signifikant höheren Prozentsatz von Patienten (62%), die den primären Wirksamkeitsendpunkt eines kombinierten biochemischen Ansprechens in Monat 12 im Vergleich zu Placebo (20%) erreichten (p < 0,0001). Der ULN für ALP wurde mit 116 U/L definiert. Der ULN für Gesamtbilirubin wurde mit 1,1 mg/dL definiert. Tabelle 2 zeigt die Ergebnisse nach 12 Monaten für den Prozentsatz der Patienten, die ein biochemisches Ansprechen, jede Komponente des biochemischen Ansprechens und eine ALP-Normalisierung erreichten. Insgesamt hatten 87% der Patienten bei Studienbeginn eine TB-Konzentration von weniger als oder gleich ULN. Daher war die Verbesserung des ALP der Hauptfaktor für die Ergebnisse der biochemischen Ansprechrate nach 12 Monaten.
Tabelle 2: Studie CB8025-32048: Kombinierter biochemischer Endpunkt und ALP-Normalisierung mit Lyvdelzi mit oder ohne UDCA
Lyvdelzi 10 mg Placebo(n=65) Differenz zwischen den
(n=128) Behandlungen % (95%-KI)d
Primärer kombinierter
Endpunkt in Monat 12
Biochemische Ansprechrate, 79 (62) [53; 70] 13 (20) [10; 30] 42 (28; 53) p < 0,0001
n (%)a, b [95%-KI]
Komponenten des biochemisch
en Ansprechens
ALP < 1,67 × ULN, n (%) 84 (66) 17 (26) 39 (25; 52)
ALP-Verringerung um 107 (84) 21 (32) 51 (37; 63)
mindestens 15%, n (%)
Gesamtbilirubin ≤ ULN, n 104 (81) 50 (77) 4 (-7; 17)
(%)
ALP-Normalisierung, n 32 (25) [18; 33] 0 (0) [0; 0] 25 (18; 33) p < 0,0001
(%)b, c [95%-KI]
Patienten, welche die Behandlung vor Monat 12 abbrachen oder Patienten mit fehlenden Daten wurden als Non-Responder eingestuft.
a Biochemisches Ansprechen definiert als ALP-Wert < 1,67 × ULN, ALP-Abnahme ≥15% und Gesamtbilirubin ≤ ULN.
b Berechnung der p-Werte anhand des Cochran-Mantel-Haenszel-Tests, stratifiziert nach ALP-Spiegel zu Studienbeginn (< 350 E/l versus ≥350 E/l) und Pruritus-Score auf der numerischen Rating-Skala (NRS) zu Studienbeginn (< 4 versus ≥4).
c ALP-Normalisierung definiert als ALP ≤ ULN.
d Die 95%-Konfidenzintervalle (KI) wurden basierend auf der Methode von Miettinen und Nurminen ohne Stratifizierung angegeben.
Biochemische Leberwerte
Abbildung 1 zeigt die mittleren ALP-Verringerungen bei mit Lyvdelzi behandelten Patienten im Vergleich zu Placebo über 12 Monate. Verringerungen wurden in Monat 1 beobachtet, setzten sich bis Monat 6 fort und hielten bis Monat 12 an. Die mittlere Veränderung (SE) der kleinsten Quadrate (LS) gegenüber dem Ausgangswert der ALP in Monat 12 betrug in der Gruppe mit Lyvdelzi 10 mg -134 (-151, -117) E/l und in der Placebo-Gruppe -17 (-40, 6) E/l.
Abbildung 1. ALP-Veränderung ab Studienbeginn über 12 Monate in der Studie CB8025-32048 nach Behandlungsarm mit oder ohne UDCAa
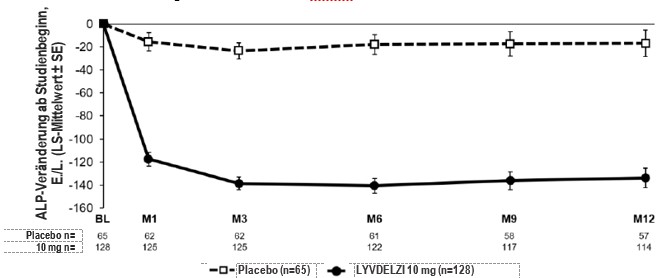
a In der Studie CB8025-32048 hatten 12 Patienten (6%) eine Unverträglichkeit gegenüber UDCA und erhielten die Behandlung als Monotherapie: 8 Patienten (6%) im Arm mit LYVDELZI 10 mg und 4 Patienten (6%) im Placebo-Arm.
In der Subgruppe von Patienten mit ALP < 350 E/l zu Studienbeginn erreichten 76% (71/93) bzw. 23% (11/47) Patienten im Arm mit Lyvdelzi 10 mg bzw. im Placebo-Arm in Monat 12 ein Ansprechen. Von den Patienten mit ALP ≥350 E/l zu Studienbeginn erreichten 23% (8/35) bzw. 11% (2/18) im Arm mit Lyvdelzi 10 mg bzw. im Placebo-Arm in Monat 12 ein Ansprechen.
Pruritus
Bei Patienten mit durchschnittlichen Pruritus-Scores von ≥4 zu Studienbeginn, gemessen anhand des Pruritus-NRS-Scores, führte Lyvdelzi im Vergleich zu Placebo in Monat 6 zu einer signifikanten Verringerung des Pruritus, einem sekundären Schlüssel-Endpunkt in der Studie CB8025-32048 (siehe Tabelle 3). Ein einzelner, von Patienten gemeldeter Endpunkt, die Numerische Rating-Skala (NRS) für Pruritus, bewertete in Studie CB8025-32048 die täglich schlimmste Juckreizintensität der Patienten auf einer 11-stufigen Bewertungsskala mit Werten von 0 ( "kein Juckreiz" ) bis 10 ( "schlimmster vorstellbarer Juckreiz" ). Die NRS für Pruritus wurde täglich in einer 14-tägigen Anlaufphase vor der Randomisierung bis zum 6. Monat durchgeführt.
Tabelle 3 zeigt die Ergebnisse des Vergleichs zwischen Lyvdelzi und Placebo bei diesem Endpunkt, der die Veränderung des Pruritus-Scores gegenüber dem Ausgangswert nach 6 Monaten bei Patienten mit einem durchschnittlichen Pruritus-Score von mindestens 4 zu Studienbeginn bewertet. Der durchschnittliche Pruritus-Score zu Studienbeginn für jeden Patienten wurde durch Mittelwertbildung der Pruritus-NRS-Scores berechnet, die in der Anlaufphase und an Tag 1 vor Behandlungsbeginn erhoben wurden. Der Pruritus-Score nach 6 Monaten wurden für jeden Patienten durch Mittelwertbildung der Pruritus-NRS-Scores innerhalb der letzten Woche des Monats berechnet. Bei den mit Lyvdelzi behandelten Patienten wurde eine grössere Verbesserung des Pruritus im Vergleich zu Placebo festgestellt.
Tabelle 3: Veränderung des Pruritus-Scores von Studienbeginn bis Monat 6 bei PBC-Patienten in der Studie CB8025-32048 mit einem durchschnittlichen Pruritus-Score von ≥4 zu Studienbeginna
Lyvdelzi 10 mg Placebo(n=23) Differenz zwischen den
(n=49) Behandlungen % (95%-KI)
Durchschnittlicher 6,1 (1,4) 6,6 (1,4) -
Pruritus-Score zu
Studienbeginn, Mittelwert
(SD)b
Veränderung des Pruritus-Sc
ores von Studienbeginn bis
Monat 6c
Mittelwert (SE) -3,2 (0,28) -1,7 (0,41) -1,5 (-2,5; -0,5) p <0,005
a Bewertet anhand der Pruritus-NRS, mit der die Intensität des täglichen Juckreizes der Patienten in seiner schlimmsten Ausprägung auf einer 11-Punkte-Skala von 0 ( "Kein Juckreiz" ) bis 10 ( "Schlimmster vorstellbarer Juckreiz" ) beurteilt wird. Die Pruritus-NRS wurde im Rahmen einer ≥14-tägigen Anlaufphase vor der Randomisierung bis Monat 6 täglich ermittelt. Mittelschwerer bis schwerer Pruritus war definiert als Pruritus-NRS-Score ≥4.
b Der Wert zu Studienbeginn schloss den Mittelwert aller während der Anlaufphase und an Tag 1 täglich berichteten Scores ein. Die Pruritus-Scores wurden für jeden Patienten für die Monate nach Studienbeginn durch Mittelung der Pruritus-NRS-Scores innerhalb der für jeden Monat vorgesehenen Woche berechnet.
c Basierend auf den LS-Mittelwerten aus einem gemischten Modell mit Messwiederholungen (Mixed-Effect Model for Repeated Measures, MMRM) für die Veränderung gegenüber Studienbeginn in den Monaten 1 (Woche 4), 3 (Woche 12) und 6 (Woche 26) unter Berücksichtigung des durchschnittlichen Pruritus-Scores zu Studienbeginn, des ALP-Spiegels zu Studienbeginn (< 350 E/L versus ALP-Spiegel ≥350 E/L), des Behandlungsarms, der Zeit (in Monaten) und der Interaktion zwischen Behandlung und Zeit.
|