Eigenschaften/WirkungenATC-Code
L04AA58
Wirkungsmechanismus
Efgartigimod alfa ist ein Fragment des humanen IgG1-Antikörpers, dessen Affinität für den neonatalen Fc-Rezeptor (FcRn) erhöht wurde. Efgartigimod alfa bindet an FcRn, was zu einer Verringerung der Spiegel von zirkulierendem IgG, einschliesslich pathogener IgG-Autoantikörper, führt. Efgartigimod alfa beeinflusst weder die Spiegel anderer Immunglobuline (IgA, IgD, IgE oder IgM) noch senkt es den Albuminspiegel.
IgG-Autoantikörper sind die zugrunde liegende Ursache der Pathogenese von IgG-vermittelten Autoimmunerkrankungen.
Bei MG beeinträchtigen sie die neuromuskuläre Übertragung, indem sie an Acetylcholin-Rezeptoren (AChR), muskelspezifische Tyrosinkinase (MuSK) oder an das Low-Density-Lipoprotein-Rezeptor-verwandte Protein 4 (LRP4) binden.
Bei CIDP weisen mehrere Evidenzlinien auf die Schlüsselrolle von IgG-Autoantikörpern bei der Pathogenese dieser Krankheit hin. Dazu gehören der Nachweis autoreaktiver IgG-Antikörper gegen Bestandteile myelinisierter Nerven, die passive Übertragung von CIDP-Symptomen auf Tiermodelle mittels Seren oder IgG von CIDP-Patienten sowie die therapeutische Wirkung von Plasmaaustausch und Immunadsorption bei der Behandlung von Patienten mit CIDP.
Pharmakodynamik
Intravenöse Darreichungsform
In der doppelblinden, placebokontrollierten Studie ARGX-113-1704 bei gMG-Patienten verringerte Efgartigimod alfa bei der empfohlenen Dosis von 10 mg/kg und dem empfohlenen Behandlungsplan (einmal wöchentliche Anwendung über 4 Wochen) die IgG-Spiegel und die AChR-Autoantikörperspiegel (AChR-Ab) im Serum (siehe Rubrik "Dosierung/Anwendung" ). Die mittlere prozentuale Abnahme des IgG-Gesamtspiegels im Vergleich zum Ausgangswert erreichte eine Woche nach der letzten Infusion des ersten Behandlungszyklus einen Maximalwert von 61 % und hatte 9 Wochen nach der letzten Infusion wieder den Ausgangswert erreicht. Eine ähnliche Wirkung wurde bei allen IgG-Subtypen beobachtet. Die Abnahme der AChR-Autoantikörperspiegel folgte einem ähnlichen Zeitverlauf mit einer maximalen mittleren prozentualen Abnahme von 58 % eine Woche nach der letzten Infusion und einer Wiederherstellung des Ausgangswerts 7 Wochen nach der letzten Infusion. Ähnliche Veränderungen wurden im zweiten Zyklus der Studie festgestellt.
Subkutane Darreichungsform
In der Studie ARGX-113-2001 folgte die Abnahme der AChR-Ab-Spiegel einem vergleichbaren Zeitverlauf wie der IgG-Gesamtspiegel und war in den mit Efgartigimod alfa subkutan und intravenös behandelten Gruppen ähnlich. In der mit Efgartigimod alfa subkutan behandelten Gruppe und in der mit Efgartigimod alfa intravenös behandelten Gruppe wurde eine Woche nach der letzten Verabreichung eine maximale mittlere prozentuale Abnahme der AChR-Ab-Spiegel von 62,2 % bzw. 59,6 % beobachtet. Sowohl in der mit Efgartigimod alfa subkutan behandelten Gruppe als auch in der mit Efgartigimod alfa intravenös behandelten Gruppe war eine Abnahme des IgG-Gesamtspiegels und des AChR-Ab-Spiegels mit einem klinischen Ansprechen verbunden, gemessen anhand der Veränderung des MG-ADL-Gesamtscores gegenüber Baseline (siehe Abbildung 1).
Abbildung 1. Zusammenhang zwischen Gesamt-IgG und AChR-Ab und MG-ADL-Gesamtscore in der AChR-Ab-seropositiven Population unter Behandlung mit Efgartigimod alfa subkutan (1A) und Efgartigimod alfa intravenös (1B) (Studie ARGX-113-2001)
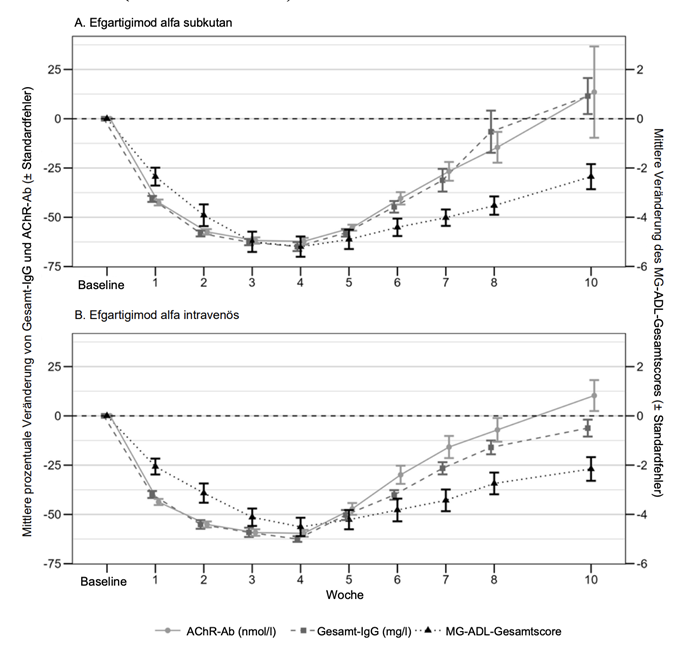
In der Studie ARGX-113-1802 bei Patienten mit CIDP, die eine kontinuierliche, einmal wöchentliche Anwendung von 1000 mg Efgartigimod alfa subkutan erhielten, blieb die mittlere prozentuale Veränderung des Gesamt-IgG-Spiegels gegenüber Baseline ab Woche 4 während des gesamten Behandlungszeitraums erhalten (die mittlere prozentuale Verringerung gegenüber Baseline lag zwischen 66,8 und 71,6 %).
Klinische Wirksamkeit
Generalisierte Myasthenia gravis
Intravenöse Darreichungsform
Die Wirksamkeit von Efgartigimod alfa zur Behandlung von Erwachsenen mit generalisierter Myasthenia gravis (gMG) wurde in einer 26-wöchigen, multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (ARGX-113-1704) untersucht.
In dieser Studie mussten die Patienten beim Screening die folgenden Hauptkriterien erfüllen:
-Klasse II, III oder IV gemäss der Klassifikation der Myasthenia Gravis Foundation of America (MGFA);
-Patienten mit positiven oder negativen serologischen Tests auf Antikörper gegen AChR;
-Gesamtscore von ≥5 im MG-ADL (MG-Activities of Daily Living);
-Behandlung mit stabil eingestellten Dosen einer MG-Therapie vor dem Screening, einschliesslich Acetylcholinesterase (AChE)-Hemmer, Steroiden oder einer nichtsteroidalen immunsuppressiven Therapie (NSIST), entweder in Kombination oder als Monotherapie [NSISTs schlossen unter anderem Azathioprin, Methotrexat, Cyclosporin, Tacrolimus, Mycophenolatmofetil und Cyclophosphamid ein];
-IgG-Werte von mindestens 6 g/l.
Patienten mit gMG der MGFA-Klasse V; Patienten mit dokumentiertem unzureichendem klinischem Ansprechen auf Plasmapherese (plasma exchange, PLEX), Patienten, die einen Monat vor Behandlungsbeginn mit PLEX, intravenösem Immunglobulin (IVIg) und sechs Monate vor Behandlungsbeginn mit monoklonalen Antikörpern behandelt wurden, Patienten, die in den letzten drei Monaten vor Studienbeginn eine Thymektomie erhalten hatten und Patienten mit aktiver (akuter oder chronischer) Hepatitis-B-Infektion, mit Hepatitis-C-Seropositivität, mit AIDS-Diagnose, oder einer schweren Infektion in den letzten 8 Wochen oder einer nicht erfolgreich behandelten malignen Grunderkrankung in den letzten drei Jahren einschliesslich malignem Thymom waren von der Teilnahme an den Studien ausgeschlossen.
Es wurden insgesamt 167 Patienten in die Studie aufgenommen und erhielten nach Randomisierung entweder Efgartigimod alfa intravenös (n = 84) oder Placebo (n = 83). Die Behandlungsgruppen wiesen ähnliche Baseline-Merkmale auf, einschliesslich des medianen Alters bei der Diagnose [45 (19-81) Jahre], des Geschlechts [die meisten waren weiblich; 75 % (Efgartigimod alfa) vs. 66 % (Placebo)], der ethnischen Zugehörigkeit [die meisten Patienten waren weiss; 84,4 %] und des medianen Zeitraums seit der Diagnose [8,2 Jahre (Efgartigimod alfa) und 6,9 Jahre (Placebo)].
Die meisten Patienten (77 % in jeder Gruppe) wurden positiv auf Antikörper gegen AChR (AChR-Ab) getestet, und 23 % der Patienten wurden negativ auf AChR-Ab getestet.
Während der Studie erhielten über 80 % der Patienten in jeder Gruppe AChE-Hemmer, über 70 % in jeder Behandlungsgruppe erhielten Steroide und etwa 60 % in jeder Behandlungsgruppe erhielten eine nichtsteroidale immunsuppressive Therapie (NSIST) in stabiler Dosierung. Zu Beginn der Studie hatten etwa 30 % der Patienten in jeder Behandlungsgruppe vorher noch keine NSIST erhalten.
Der mediane MG-ADL-Gesamtscore betrug in beiden Behandlungsgruppen 9,0, und der mediane Quantitative Myasthenia Gravis(QMG)-Score betrug 17 in der mit Efgartigimod alfa behandelten Gruppe und 16 in der Placebo-Gruppe.
Die Behandlung der Patienten mit Efgartigimod alfa intravenös erfolgte im Rahmen des empfohlenen Dosierungsschemas von 10 mg/kg einmal wöchentlich über 4 Wochen und mit maximal 3 Behandlungszyklen (siehe Rubrik "Dosierung/Anwendung" ).
Die Wirksamkeit von Efgartigimod alfa wurde anhand der Skala MG-ADL (Myasthenia Gravis-Specific Activities of Daily Living) gemessen, mit der die Auswirkungen der gMG auf tägliche Aktivitäten beurteilt wird. Der Gesamtscore liegt im Bereich von 0 bis 24, wobei höhere Werte eine stärkere Beeinträchtigung bedeuten. In dieser Studie war ein MG-ADL-Responder ein Patient mit einer Verringerung des MG-ADL-Gesamtscores um ≥2 Punkte in mindestens 4 aufeinanderfolgenden Wochen im Vergleich zum Wert zu Anfang des Behandlungszyklus, wobei die erste Verringerung nicht später als eine Woche nach der letzten Infusion des Zyklus festzustellen war.
Die Wirksamkeit von Efgartigimod alfa wurde auch anhand des QMG-Scores gemessen, einem System zur Einstufung der Muskelschwäche mit einem möglichen Gesamtscore von 0 bis 39, bei dem höhere Scores eine stärkere Beeinträchtigung bedeuten. In dieser Studie war ein QMG-Responder ein Patient mit einer Verringerung des QMG-Scores um ≥3 Punkte in mindestens 4 aufeinanderfolgenden Wochen im Vergleich zum Wert zu Anfang des Behandlungszyklus, wobei die erste Verringerung nicht später als eine Woche nach der letzten Infusion des Zyklus festzustellen war.
Der primäre Wirksamkeitsendpunkt war der Vergleich des Prozentsatzes der MG-ADL-Responder während des ersten Behandlungszyklus (C1) zwischen den Behandlungsgruppen in der Population der AChR-Ab-seropositiven Patienten.
Ein wichtiger sekundärer Endpunkt war der Vergleich des Prozentsatzes der QMG-Responder während C1 zwischen beiden Behandlungsgruppen bei den AChR-Ab-seropositiven Patienten.
Tabelle 2. MG-ADL- und QMG-Responder in Zyklus 1 in der Population der AChR-Ab-seropositiven Patienten (mITT-Analysegruppe)
Population Efgartigimod alfan/N Placebon/N (%) P-Wert Differenz Efgartigim
(%) od alfa - Placebo
(95 %-KI)
MG-ADL AChR-Ab-seropositiv 44/65 (67,7) 19/64 (29,7) <0,0001 38,0 (22,1; 54,0)
QMG AChR-Ab-seropositiv 41/65 (63,1) 9/64 (14,1) <0,0001 49,0 (34,5; 63,5)
AChR-Ab = Anti-Acetylcholin-Rezeptor-Antikörper; MG-ADL = Myasthenia Gravis Activities of Daily Living (Fragebogen zur Beurteilung der MG-Krankheitsaktivität); QMG = Quantitative Myasthenia Gravis (Fragebogen zur Bestimmung eines Quantitativen Myasthenia Gravis-Score); mITT = modifizierte Intent-to-treat-Gruppe; n = Anzahl der Patienten, bei denen die jeweilige Beobachtung gemacht wurde; N = Anzahl der Patienten in der Analysegruppe; KI = Konfidenzintervall;
Logistische Regression, stratifiziert nach AChR-Ab-Status (sofern zutreffend), Japanisch/Nicht-Japanisch und Standardbehandlung, mit MG-ADL-/QMG-Baseline-Score als Kovariate
Zweiseitiger exakter p-Wert
Analysen zeigen, dass die MG-ADL-Responderraten während des zweiten Behandlungszyklus ähnlich waren wie im ersten Behandlungszyklus (siehe Tabelle 3).
Tabelle 3. MG-ADL- und QMG-Responder in Zyklus 2 in der Population der AChR-Ab-seropositiven Patienten (mITT-Analysegruppe)
Population Efgartigimod alfa n/N (%) Placebo n/N (%)
MG-ADL AChR-Ab-seropositiv 36/51 (70,6) 11/43 (25,6)
QMG AChR-Ab-seropositiv 24/51 (47,1) 5/43 (11,6)
AChR-Ab = Anti-Acetylcholin Rezeptor-Antikörper; MG-ADL = Myasthenia Gravis Activities of Daily Living (Fragebogen zur Beurteilung der MG-Krankheitsaktivität); QMG = Quantitative Myasthenia Gravis (Fragebogen zur Bestimmung eines Quantitativen Myasthenia Gravis-Score); mITT = modifizierte Intent-to-treat-Gruppe; n = Anzahl der Patienten, bei denen die jeweilige Beobachtung gemacht wurde; N = Anzahl der Patienten in der Analysegruppe.
Bei Patienten mit einer Thymektomie in der Vorgeschichte waren 27 (60 %) Patienten in der Efgartigimod alfa-Gruppe MG-ADL-Responder im Vergleich zu 8 (27 %) Patienten in der Placebo-Gruppe.
Exploratorische Daten zeigen, dass bei 37/44 (84 %) der mit Efgartigimod alfa intravenös behandelten Patienten der AChR-Ab-seropositiven MG-ADL-Responder ein Ansprechen innerhalb von 2 Wochen nach der ersten Infusion beobachtet wurde.
In der doppelblinden, placebokontrollierten Studie (ARGX-113-1704) konnte gemäss klinischem Studienprotokoll ein nachfolgender Behandlungszyklus erst eingeleitet werden, wenn alle folgenden Kriterien erfüllt waren:
(1) die Mindestzeit zwischen den Behandlungszyklen betrug 8 Wochen ab der ersten Infusion des vorherigen Zyklus;
(2) der Patient wies einen MG-ADL-Gesamtscore von ≥5 Punkten mit >50 % des Gesamtscores durch nicht-okuläre Symptome auf und
(3) nur für Patienten, die im vorangegangenen Behandlungszyklus den Responder Status (Definition s. oben) erreicht haben und nun einen Verlust des Ansprechens (definiert als eine Verringerung des MG-ADL-Gesamtscores <2 Punkte im Vergleich zum entsprechenden Zyklus-Ausgangs-Wert) zeigen.
In der Gesamtpopulation betrug die mittlere Zeit bis zum zweiten Behandlungszyklus in der Efgartigimod alfa intravenös behandelten Gruppe 13 Wochen (Standardabweichung 5,5 Wochen) und die mediane Zeit 10 Wochen (8-26 Wochen) ab der ersten Infusion im ersten Behandlungszyklus.
Bei Patienten, die auf die Behandlung ansprachen (Verringerung des MG-ADL-Gesamtscores um ≥2 Punkte vs Zyklus-Baseline), betrug die Dauer der klinischen Besserung bei 5/44 Patienten (11 %) 5 Wochen, bei 14/44 Patienten (32 %) 6-7 Wochen, bei 10/44 Patienten (23 %) 8-11 Wochen und bei 15/44 Patienten (34 %) 12 Wochen oder mehr.
Subkutane Darreichungsform
Es wurde eine 10-wöchige, randomisierte, offene, multizentrische Parallelgruppenstudie (ARGX-113-2001) bei erwachsenen Patienten mit gMG durchgeführt, um die Nichtunterlegenheit der pharmakodynamischen Wirkung von Efgartigimod alfa subkutan im Vergleich zu Efgartigimod alfa intravenös zu bewerten. Die wichtigsten Ein- und Ausschlusskriterien waren dieselben wie in der Studie ARGX-113-1704.
Es wurden insgesamt 110 Patienten randomisiert und erhielten über 4 Wochen einen Zyklus mit einmal wöchentlicher Verabreichung von entweder 1000 mg Efgartigimod alfa subkutan (n = 55) oder 10 mg/kg Efgartigimod alfa intravenös (n = 55). Die meisten Patienten waren positiv auf Antikörper gegen AChR (AChR-Ab): 45 Patienten (82 %) in der mit Efgartigimod alfa subkutan behandelten Gruppe und 46 Patienten (84 %) in der mit Efgartigimod alfa intravenös behandelten Gruppe. Alle Patienten erhielten vor dem Screening stabile Dosen einer MG-Therapie, die AChE-Hemmer, Steroide oder NSIST, entweder in Kombination oder als Monotherapie, umfasste.
Die Baseline-Merkmale in den Behandlungsgruppen waren vergleichbar.
Während der Studie erhielten über 80 % der Patienten in jeder Gruppe AChE-Hemmer, über 60 % der Patienten in jeder Gruppe erhielten Steroide und etwa 40 % in jeder Behandlungsgruppe erhielten NSIST in stabilen Dosen. Bei der Aufnahme in die Studie hatten etwa 56 % der Patienten in jeder Behandlungsgruppe zuvor noch keine NSIST erhalten.
Der primäre Endpunkt war der Vergleich der prozentualen Verringerung der IgG-Gesamtspiegel gegenüber dem Baseline-Wert an Tag 29 zwischen den Behandlungsgruppen in der Gesamtpopulation. Die Ergebnisse in der AChR-Ab-seropositiven Population zeigen, dass Efgartigimod alfa subkutan gegenüber Efgartigimod alfa intravenös nicht unterlegen ist (siehe Tabelle 4).
Tabelle 4. ANCOVA der prozentualen Veränderung des IgG-Gesamtspiegels gegenüber Baseline an Tag 29 in der AChR-Ab-seropositiven Population (mITT-Analysegruppe)
Efgartigimod alfa Efgartigimod alfa Differenz Efgartigim
s.c. i.v. od alfa s.c. -
Efgartigimod alfa
i.v.
N LS-Mittelwert 95 %-KI N LS-Mittelwert 95 %-KI Differenz der 95 %-KI p-Wert
LS-Mittelwerte
41 -66,9 -69,78; -64,02 43 -62,4 -65,22; -59,59 -4,5 -8,53; -0,46 <0,0001
AChR-Ab = Anti-Acetylcholin-Rezeptor-Antikörper; ANCOVA = Kovarianzanalyse; KI = Konfidenzintervall; s.c. = subkutan; i.v. = intravenös; LS = Least Squares (Kleinstquadrate); mITT = modifizierte Intent-to-treat-Analysegruppe; N = Anzahl der mittels ANCOVA analysierten Patienten
Sekundäre Wirksamkeitsendpunkte waren Vergleiche des Prozentsatzes der MG-ADL- und QMG-Responder, wie in der Studie ARGX-113-1704 definiert, zwischen beiden Behandlungsgruppen. Die Ergebnisse in der AChR-Ab-seropositiven Population sind in Tabelle 5 dargestellt.
Tabelle 5. MG-ADL- und QMG-Responder an Tag 29 in der AChR-Ab-seropositiven Population (mITT-Analysegruppe)
Efgartigimod alfa Efgartigimod alfa Differenz Efgartigimod alfa
s.c.n/N (%) i.v.n/N (%) s.c.-Efgartigimod alfa i.v. (95
%-KI)
MG-ADL 32/45 (71,1) 33/46 (71,7) -0,6 (-19,2 bis 17,9)
QMG 31/45 (68,9) 24/45 (53,3) 15,6 (-4,3 bis 35,4)
AChR-Ab = Anti-Acetylcholin-Rezeptor-Antikörper; MG-ADL = Myasthenia Gravis Activities of Daily Living (Fragebogen zur Beurteilung der MG-Krankheitsaktivität); QMG = Quantitative Myasthenia Gravis (Fragebogen zur Bestimmung eines Quantitativen Myasthenia Gravis-Scores); s.c. = subkutan; i.v. = intravenös; mITT = modifizierte Intent-to-treat-Analysegruppe; n = Anzahl der Patienten, bei denen die jeweilige Beobachtung gemacht wurde; N = Anzahl der Patienten in der Analysegruppe; KI = Konfidenzintervall;
Exploratorische Daten zeigen, dass unter den AChR-Ab-seropositiven MG-ADL-Respondern bei 28/32 Patienten (88 %), die mit Efgartigimod alfa subkutan behandelt wurden, und bei 27/33 Patienten (82 %), die mit Efgartigimod alfa intravenös behandelt wurden, innerhalb von 2 Wochen nach der ersten Verabreichung ein Ansprechen erfolgte.
Chronisch-entzündliche demyelinisierende Polyneuropathie
Die Wirksamkeit von Efgartigimod alfa subkutan zur Behandlung von Erwachsenen mit CIDP wurde in der prospektiven, multizentrischen Studie ARGX-113-1802 untersucht, die in 2 Behandlungsphasen durchgeführt wurde: eine offene Phase A und eine doppelblinde, placebokontrollierte Phase B mit randomisiertem Absetzen.
Die Patienten hatten in den letzten 6 Monaten vor Studienaufnahme entweder eine CIDP-Behandlung erhalten oder nicht. Diejenigen, die zuvor eine CIDP-Behandlung erhalten hatten, sowie diejenigen, die keine CIDP-Behandlung erhalten hatten und bei denen keine Hinweise auf eine kürzlich eingetretene Verschlechterung der CIDP vorlagen, traten in eine behandlungsfreie Vorlaufphase ein. Patienten, bei denen Hinweise auf eine klinisch bedeutsame Verschlechterung vorlagen, nahmen anschliessend an Phase A der Studie teil. Diejenigen Patienten ohne CIDP-Behandlung, bei denen vor kurzem eine Verschlechterung der CIDP nachgewiesen wurde, liessen die Vorlaufphase aus und traten direkt in Phase A ein.
Insgesamt wurden 322 Patienten in Phase A aufgenommen. Die Patienten erhielten bis zu 12 einmal wöchentliche Injektionen von Efgartigimod alfa subkutan in einer Dosierung von 1000 mg, bis bei 2 aufeinanderfolgenden Studienbesuchsterminen eine bestätigte klinische Verbesserung (ECI, evidence of clinical improvement) festgestellt wurde. Anschliessend traten die Patienten mit bestätigter ECI in Phase B der Studie ein und erhielten nach dem Zufallsprinzip entweder wöchentlich Efgartigimod alfa subkutan (111 Patienten) oder Placebo (110 Patienten). Eine ECI war definiert als klinische Verbesserung auf der Skala Adjusted Inflammatory Neuropathy Cause and Treatment (aINCAT) oder als Verbesserung auf der Inflammatory Rasch-built Overall Disability Scale (I-RODS)/Grip Strength bei Patienten, die sich vor Phase A nur auf diesen Skalen verschlechtert hatten.
In Phase A hatten die Patienten ein medianes Alter von 54 Jahren (Bereich: 20 bis 82 Jahre), einen medianen Zeitraum seit der CIDP-Diagnose von 2,8 Jahren und einen medianen INCAT-Score von 4,0. 65 Prozent waren männlich und 66 % waren weiss. In Phase B hatten die Patienten ein medianes Alter von 55 Jahren (Bereich: 20 bis 82 Jahre), einen medianen Zeitraum seit der CIDP-Diagnose von 2,2 Jahren und einen medianen INCAT-Score von 3,0. 64 Prozent waren männlich und 65 % waren weiss. Die Merkmale bei Baseline in Phase B waren zwischen den Behandlungsgruppen ähnlich.
In Phase A war der primäre Endpunkt der Prozentsatz der Responder, d.h. der Patienten, die eine bestätigte ECI erreichten. Der primäre Endpunkt wurde bei 66,5 % der Patienten erreicht; weitere Einzelheiten sind in Tabelle 6 aufgeführt.
Ein sekundärer Endpunkt in Phase A war die Zeit bis zur ersten bestätigten ECI. Woche 4 war der früheste Zeitpunkt, zu dem die Kriterien für eine ECI erfüllt werden konnten. Zu diesem Zeitpunkt erreichten bis zu 40 % der Patienten eine ECI. Auf der Grundlage einer zusätzlichen, vorab festgelegten Analyse zeigten 25 % der Patienten nach 9 Tagen eine klinisch relevante Verbesserung bei mindestens einem der 3 Parameter (aINCAT, I-RODS oder Grip Strength).
Die Mehrzahl der Patienten erreichte eine bestätigte ECI in allen vorherigen CIDP-Medikationsgruppen.
Tabelle 6. Nachweis der klinischen Verbesserung bei Patienten mit CIDP in Phase A der Studie ARGX-113-1802
ECI-Responder und Zeit bis zur ersten bestätigten ECI Phase A
Efgartigimod alfa s.c. (N = 322)
ECI-Responder (Patienten mit bestätigter klinischer Verbesserung) n/N 214/322 (66,5 %) (61,0;
(%) (95 %-KI) 71,6)
Zeit bis zur ersten bestätigten ECI in Tagen Median (95 %-KI) 43,0 (31,0; 51,0)
n = Anzahl der Patienten, für die die Beobachtung gemeldet wurde; N = Anzahl der Patienten in der Analysegruppe
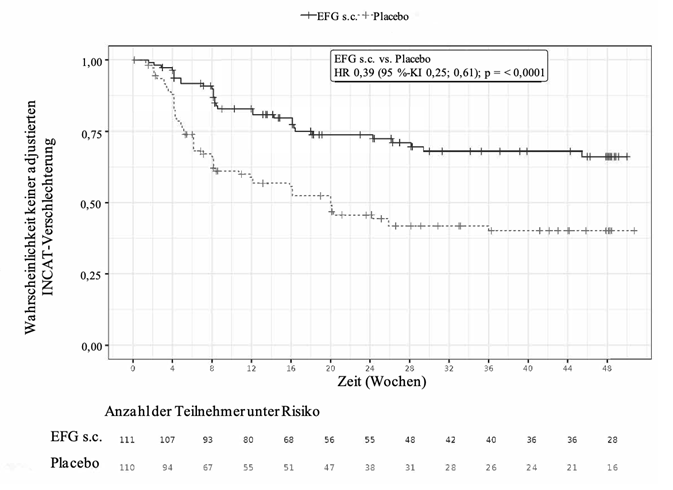
In Phase B war der primäre Endpunkt definiert als die Zeit bis zum Auftreten des ersten Nachweises einer klinischen Verschlechterung (ein Anstieg des aINCAT-Wertes um 1 Punkt im Vergleich zu Baseline von Phase B, der bei einem nachfolgenden Besuchstermin nach dem ersten Anstieg des aINCAT-Wertes um 1 Punkt bestätigt wurde, oder ein Anstieg des aINCAT-Wertes um ≥2 Punkte im Vergleich zu Baseline von Phase B). Patienten, die Efgartigimod alfa subkutan erhielten, blieben im Vergleich zu Patienten, die Placebo erhielten, signifikant länger rezidivfrei (d.h. ohne klinische Verschlechterung), was durch eine Hazard Ratio von 0,394 [95 %-KI (0,253; 0,614)] belegt wurde. Bei 31/111 Patienten (27,9 %), die in Phase B der Studie Efgartigimod alfa subkutan erhielten, kam es zu einem Rezidiv, verglichen mit 59/110 Patienten (53,6 %), die Placebo erhielten. Die Ergebnisse sind in Tabelle 7 und Abbildung 2 dargestellt.
Tabelle 7. Erster Nachweis der klinischen Verschlechterung bei Patienten mit CIDP in Phase B der Studie ARGX-113-1802
Zeit bis zum 1. aINCAT-Anstieg (klinische Phase B
Verschlechterung)
Efgartigimod alfa s.c. (N = 111) Placebo (N = 110)
Hazard Ratio (95 %-KI) 0,394 (0,253; 0,614) p-Wert
<0,0001
Mediane Zeit in Tagen (95 %-KI) NC (NC; NC) 140,0 (75,0; NC)
NC = nicht berechnet; N = Anzahl der Patienten in der Analysegruppe; aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment
Abbildung 2. Zeit bis zur ersten aINCAT-Verschlechterung (Kaplan-Meier-Kurve) bei Patienten mit CIDP in Phase B der Studie ARGX-113-1802
|