Zusammensetzung
Wirkstoffe
Nivolumab und Relatlimab sind humane Immunoglobulin G4 (IgG4) monoklonale Antikörper (HuMAb), durch rekombinante DNA Technologie hergestellt in Chinese Hamster Ovary Zellen.
Hilfsstoffe
Histidinum (aus gentechnisch veränderten/m Zuckerrüben, Mais oder Soja hergestellt), Histidini hydrochloridum monohydricum (aus gentechnisch veränderten/m Zuckerrüben, Mais oder Soja hergestellt), Saccharum (aus gentechnisch veränderten Zuckerrüben hergestellt), Acidum penteticum, Polysorbatum 80, Aqua ad iniectabilia.
Darreichungsform und Wirkstoffmenge pro Einheit
Konzentrat zur Herstellung einer Infusionslösung (i.v.; steriles Konzentrat).
Klare bis opaleszierende, farblose bis blassgelbe Flüssigkeit, die (wenige) helle Partikel enthalten kann. Die Lösung hat einen pH-Wert von etwa 5,8 und eine Osmolalität von ungefähr 310 mOsm/kg.
Jeder ml Konzentrat enthält 12 mg Nivolumab und 4 mg Relatlimab.
Eine Durchstechflasche von 20 ml enthält 240 mg Nivolumab und 80 mg Relatlimab.
Indikationen/Anwendungsmöglichkeiten
Opdualag ist für die Erstlinienbehandlung von Erwachsenen mit nicht resezierbarem oder metastasiertem Melanom indiziert (siehe "Eigenschaften/Wirkungen" ).
Dosierung/Anwendung
Die Behandlung muss von Ärzten mit Erfahrung in der Behandlung von Krebs eingeleitet und überwacht werden.
Übliche Dosierung
Die empfohlene Dosierung von Opdualag ist:
-Erwachsene Patienten: Opdualag (480 mg Nivolumab und 160 mg Relatlimab) alle 4 Wochen verabreicht als intravenöse Infusion über 30 Minuten.
Therapiedauer
Die Behandlung mit Opdualag sollte so lange fortgesetzt werden, wie ein klinischer Nutzen beobachtet wird oder bis die Behandlung vom Patienten nicht mehr toleriert wird.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Eine Erhöhung oder Reduktion der Dosis wird nicht empfohlen. Eine Verzögerung der Dosierung oder ein Absetzen kann basierend auf der individuellen Sicherheit und Verträglichkeit erforderlich sein. Richtlinien für das dauerhafte Absetzen oder das Aufschieben von Dosen sind in Tabelle 1 beschrieben. Ausführliche Richtlinien zum Umgang mit immunvermittelten unerwünschten Wirkungen sind in der Rubrik "Warnhinweise und Vorsichtsmassnahmen" beschrieben.
Tabelle 1: Empfohlene Behandlungsumste
llungen für Opdualag
Immunvermittelte unerwünschte Wirkung Schweregrad Behandlungsumstellung
Immunvermittelte Pneumonitis Pneumonitis Grad 2 Die Verabreichung aufschieben bis
die Symptome abklingen, sich
auffällige Röntgenbefunde verbessern
und die Behandlung mit Kortikosteroid
en abgeschlossen ist
Pneumonitis Grad 3 oder 4 Die Behandlung
dauerhaft absetzen
Immunvermittelte Kolitis Diarrhö oder Kolitis Die Verabreichung aufschieben bis
Grad 2 oder 3 die Symptome abklingen und die
Behandlung mit Kortikosteroiden
abgeschlossen ist
Diarrhö oder Kolitis Grad 4 Die Behandlung
dauerhaft absetzen
Immunvermittelte Hepatitis AST/ALT-Erhöhung Die Verabreichung aufschieben bis
auf mehr als das 3- die Laborwerte wieder die
und bis zum 5-fachen ursprünglichen Spiegel erreichen
des ULN oder und, falls erforderlich, die
Erhöhung des Gesamt- Behandlung mit Kortikosteroiden
Bilirubins auf mehr abgeschlossen ist
als das 1,5- und
bis zum 3-fachen
des ULN
AST- oder ALT-Erhöhung auf mehr als Die Behandlung
das 5-fache des ULN, ungeachtet des dauerhaft absetzen
ursprünglichen Spiegels oder Erhöhung
des Gesamt-Bilirubins auf mehr als
das 3-fache des ULN oder
gleichzeitige AST- oder ALT-Erhöhung
auf mehr als das 3-fache des ULN und
Erhöhung des Gesamt-Bilirubins auf
mehr als das 2-fache des ULN
Immunvermittelte Nephritis und Kreatinin-Erhöhung Die Verabreichung aufschieben bis
Nierenfunktionsstörung Grad 2 oder 3 der Kreatinin-Spiegel wieder den
ursprünglichen Spiegel erreicht und
die Behandlung mit Kortikosteroiden
abgeschlossen ist
Kreatinin-Erhöhung Grad 4 Die Behandlung
dauerhaft absetzen
Immunvermittelte Endokrinopathien Symptomatische Grad Die Verabreichung aufschieben bis
2 oder 3 Hypothyreos die Symptome abklingen und die
e, Hyperthyreose, Behandlung mit Kortikosteroiden
Hypophysitis Nebenni (falls zur Behandlung der Symptome
ereninsuffizienz einer akuten Entzündung
Grad 2 Diabetes erforderlich) abgeschlossen ist. Die
Grad 3 Behandlung sollte während einer
Hormonersatztherapiea fortgesetzt
werden, soweit keine Symptome
auftreten
Hypothyreose Grad 4 Hyperthyreose Die Behandlung
Grad 4 Hypophysitis Grad 4 dauerhaft absetzen
Nebenniereninsuffizienz Grad 3 oder 4
Diabetes Grad 4
Immunvermittelte Hautreaktionen Hautausschlag Grad 3 Die Verabreichung aufschieben bis
die Symptome abklingen und die
Behandlung mit Kortikosteroiden
abgeschlossen ist
Verdacht auf Stevens-Johnson Syndrom Die Verabreichung
(SJS) oder toxischer epidermaler aufschieben
Nekrolyse (TEN)
Hautausschlag Grad 4 SJS/TEN bestätigt Die Behandlung
dauerhaft absetzen
(siehe "Warnhinweise
und Vorsichtsmassna
hmen" )
Immunvermittelte Myokarditis Myokarditis Grad 2 Die Verabreichung aufschieben bis
die Symptome abklingen und die
Behandlung mit Kortikosteroiden
abgeschlossen ist. Eine
Wiederaufnahme der Behandlung kann
nach Erholung in Betracht gezogen
werden.b
Myokarditis Grad 3 oder 4 Die Behandlung
dauerhaft absetzen
Immunvermittelte Myelitis Alle Grade Die Behandlung dauerhaft absetzen
Andere immunvermittelte unerwünschte Grad 3 (erstes Die Verabreichung aufschieben
Wirkungen Auftreten)
Grad 4 oder wiederauftretende Grad 3; Die Behandlung
andauernde Grad 2 oder 3 trotz dauerhaft absetzen
Behandlungsanpassung; wenn die
Kortikosteroid-Dosis nicht auf
täglich 10 mg Prednison oder
äquivalente Dosis eines anderen
Kortikosteroids reduziert werden kann
Hinweis: Die Toxizitätsgrade entsprechen den National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (NCI-CTCAE v5).
a Empfehlungen für die Anwendung einer Hormonersatztherapie finden Sie in der Rubrik "Warnhinweise und Vorsichtsmassnahmen" .
b Die Sicherheit einer Wiederaufnahme der Behandlung mit Opdualag bei Patienten mit einer vorangegangenen immunvermittelten Myokarditis ist nicht bekannt.
Patienten mit Leberfunktionsstörungen
Basierend auf den Ergebnissen der Populations-Pharmakokinetik (PK) ist bei Patienten mit leichter oder mässiger Leberfunktionsstörung keine Dosisanpassung erforderlich (siehe "Pharmakokinetik" ). Die Datenlage bei Patienten mit schwerer Leberfunktionsstörung ist zu begrenzt, um Schlussfolgerungen bei dieser Population ziehen zu können. Opdualag muss bei Patienten mit Leberfunktionsstörung mit Vorsicht verabreicht werden.
Patienten mit Nierenfunktionsstörungen
Basierend auf den Ergebnissen der Populations-PK ist bei Patienten mit leichter oder moderater Nierenfunktionsstörung keine Dosisanpassung erforderlich (siehe "Pharmakokinetik" ). Die Datenlage bei Patienten mit schwerer Nierenfunktionsstörung ist zu begrenzt, um Schlussfolgerungen bei dieser Population ziehen zu können. Opdualag muss bei Patienten mit Nierenfunktionsstörung mit Vorsicht verabreicht werden.
Ältere Patienten
Für ältere Patienten (≥65 Jahre) ist keine Dosisanpassung erforderlich (siehe "Pharmakokinetik" ).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Opdualag bei Kindern und Jugendlichen ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Opdualag ist nur zur intravenösen Verabreichung bestimmt. Es wird als intravenöse Infusion über einen Zeitraum von ungefähr 30 Minuten verabreicht. Die Infusion muss durch einen sterilen, nichtpyrogenen In-Line-Filter mit geringer Proteinbindung und einer Porengrösse von 0,2-1,2 µm verabreicht werden.
Opdualag darf nicht als intravenöse Druck- oder Bolus-Injektion verabreicht werden.
Opdualag kann mit Natriumchloridlösung 9 mg/ml (0,9%) zur Injektion oder Glukoselösung 50 mg/ml (5%) zur Injektion verdünnt werden (siehe "Sonstige Hinweise" ).
Anweisungen zur Handhabung des Arzneimittels vor der Anwendung, sind in der Rubrik "Sonstige Hinweise" zu finden.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe aufgelistet unter "Zusammensetzung" .
Warnhinweise und Vorsichtsmassnahmen
Immunvermittelte unerwünschte Wirkungen
Immunvermittelte unerwünschte Wirkungen können mit Nivolumab in Kombination mit Relatlimab auftreten. Die meisten immunvermittelten unerwünschten Wirkungen konnten durch entsprechende Behandlung, einschliesslich Verabreichung von Kortikosteroiden und Dosierungsanpassungen, im Schweregrad reduziert oder behoben werden (siehe "Dosierung/Anwendung" ). Immunvermittelte unerwünschte Wirkungen, die mehr als ein Körpersystem betreffen, können gleichzeitig auftreten.
Die Patienten sollten kontinuierlich überwacht (für mindestens 5 Monate nach der letzten Dosis) werden, da eine unerwünschte Wirkung von Opdualag jederzeit während oder nach dem Absetzen der Therapie auftreten kann.
Bei Verdacht auf immunvermittelte unerwünschte Wirkungen sollte eine angemessene Beurteilung erfolgen, um die Ätiologie zu bestätigen oder andere Ursachen auszuschliessen. Basierend auf dem Schweregrad der unerwünschten Wirkung sollten die Behandlungen mit Opdualag aufgeschoben und Kortikosteroide verabreicht werden. Wenn Immunsuppression mit Kortikosteroiden zur Behandlung einer unerwünschten Wirkung verwendet wird, sollte bei Einsetzen einer Verbesserung ein Ausschleichen über mindestens 1 Monat eingeleitet werden. Eine schnelle Reduzierung kann zu einer Verschlechterung oder einem Wiederauftreten der unerwünschten Wirkung führen. Eine immunsuppressive Therapie ohne Kortikosteroide sollte hinzugefügt werden, wenn trotz Kortikosteroid-Anwendung eine Verschlechterung oder keine Besserung eintritt.
Die Behandlung mit Opdualag sollte nicht wieder aufgenommen werden, während der Patient immunsuppressive Kortikosteroiddosen oder eine andere immunsuppressive Therapie erhält. Antibiotika-Prophylaxe sollte bei Patienten unter einer immunsuppressiven Therapie verwendet werden, um opportunistische Infektionen zu vermeiden.
Die Behandlung mit Opdualag muss bei Wiederauftreten von schweren immunvermittelten unerwünschten Wirkungen und bei lebensbedrohlichen immunvermittelten unerwünschten Wirkungen dauerhaft abgesetzt werden.
Immunvermittelte Pneumonitis
Schwere Pneumonitis oder interstitielle Lungenerkrankung, einschliesslich einem Fall mit Todesfolge, wurde unter Behandlung mit Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ). Die Patienten sollten auf Anzeichen und Symptome einer Pneumonitis, wie radiologische Veränderungen (z.B. fokale milchglasartige Dichteanhebung, unregelmässige Infiltrate), Dyspnoe und Hypoxie überwacht werden. Infektiöse und krankheitsbedingte Ätiologien sollten ausgeschlossen werden.
Bei Pneumonitis Grad 3 oder 4 muss Opdualag dauerhaft abgesetzt werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 2 bis 4 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden.
Bei einer (symptomatischen) Pneumonitis Grad 2 sollte Opdualag aufgeschoben und eine Behandlung mit Kortikosteroiden mit einer Dosis von 1 mg/kg/Tag Methylprednisolon-Äquivalent eingeleitet werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids wieder aufgenommen werden. Wenn trotz der Einleitung einer Behandlung mit Kortikosteroiden eine Verschlechterung oder keine Verbesserung eintritt, sollte die Kortikosteroiddosis auf 2 bis 4 mg/kg/Tag Methylprednisolon-Äquivalent erhöht werden. Opdualag muss in diesem Fall dauerhaft abgesetzt werden.
Immunvermittelte Kolitis
Schwere Diarrhö oder Kolitis wurde bei der Behandlung mit Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ). Die Patienten sollten auf Durchfall und zusätzliche Symptome einer Kolitis wie Bauchschmerzen und Schleim und/oder Blut im Stuhl überwacht werden. Cytomegalovirus (CMV) Infektion/Reaktivierung wurde bei Patienten mit Kortikosteroid-refraktärer immunvermittelter Kolitis berichtet. Infektiöse und andere Ätiologien für Diarrhö sollten ausgeschlossen werden. Deshalb müssen angemessene Laboruntersuchungen und zusätzliche Untersuchungen durchgeführt werden. Sollte sich der Verdacht auf eine Kortikosteroid-refraktäre immunvermittelte Kolitis bestätigen, dann sollte zusätzlich zur Kortikosteroid-Therapie, eine Therapie mit einem alternativen Immunsuppressivum, oder das Ersetzen der Kortikosteroid-Therapie in Betracht gezogen werden.
Bei Diarrhö oder Kolitis Grad 4 muss Opdualag dauerhaft abgesetzt werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden.
Bei Diarrhö oder Kolitis Grad 3 sollte Opdualag aufgeschoben und eine Behandlung mit Kortikosteroiden mit einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent eingeleitet werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids wieder aufgenommen werden. Wenn trotz der Einleitung einer Behandlung mit Kortikosteroiden eine Verschlechterung oder keine Verbesserung eintritt, muss Opdualag dauerhaft abgesetzt werden.
Bei Diarrhö oder Kolitis Grad 2 sollte Opdualag aufgeschoben werden. Eine anhaltende Diarrhö oder Kolitis sollte mit Kortikosteroiden mit einer Dosis von 0,5 bis 1 mg/kg/Tag Methylprednisolon-Äquivalent behandelt werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids, falls erforderlich, wieder aufgenommen werden. Wenn trotz der Einleitung einer Behandlung mit Kortikosteroiden eine Verschlechterung oder keine Verbesserung eintritt, sollte die Kortikosteroiddosis auf 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent erhöht werden. In diesem Fall muss Opdualag dauerhaft abgesetzt werden.
Immunvermittelte Hepatitis
Schwere Hepatitis wurde bei der Behandlung mit Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ). Die Patienten sollten auf Anzeichen und Symptome einer Hepatitis wie erhöhte Transaminasewerte und Gesamt-Bilirubinspiegel überwacht werden. Infektiöse und krankheitsbedingte Ätiologien sollten ausgeschlossen werden.
Bei einem AST- oder ALT-Anstieg auf mehr als das 5-fache des ULN unabhängig vom Ausgangswert, oder bei einem Gesamt-Bilirubin-Anstieg auf mehr als das 3-fache des ULN, oder bei gleichzeitigem AST- oder ALT-Anstieg auf mehr als das 3-fache des ULN und einem Gesamt-Bilirubin-Anstieg auf mehr als das 2-fache des ULN muss Opdualag dauerhaft abgesetzt werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden.
Bei einem AST- oder ALT-Anstieg auf mehr als das 3-fache bis zum 5-fachen des ULN, oder bei einem Gesamt-Bilirubin-Anstieg auf mehr als das 1,5-fache bis zum 3-fachen des ULN sollte Opdualag aufgeschoben werden. Anhaltende Erhöhungen dieser Laborwerte sollten mit Kortikosteroiden mit einer Dosis von 0,5 bis 1 mg/kg/Tag Methylprednisolon-Äquivalent behandelt werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids, falls erforderlich, wieder aufgenommen werden. Wenn trotz der Einleitung einer Behandlung mit Kortikosteroiden eine Verschlechterung oder keine Verbesserung eintritt, sollte die Kortikosteroiddosis auf 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent erhöht werden. Opdualag muss in diesem Fall dauerhaft abgesetzt werden.
Immunvermittelte Nephritis oder Nierenfunktionsstörung
Schwere Nephritis und Nierenfunktionsstörung wurden bei der Behandlung mit Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ). Die Patienten sollten auf Anzeichen und Symptome einer Nephritis und Nierenfunktionsstörung überwacht werden. Bei den meisten Patienten kommt es zu einem asymptomatischen Anstieg des Serum-Kreatinins. Krankheitsbedingte Ätiologien sollten ausgeschlossen werden.
Bei einem erhöhten Serum-Kreatinin-Spiegel Grad 4 muss Opdualag dauerhaft abgesetzt werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden.
Bei einem erhöhten Serum-Kreatinin-Spiegel Grad 2 oder 3 sollte die Behandlung mit Opdualag aufgeschoben werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 0,5 bis 1 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids wieder aufgenommen werden. Wenn trotz der Einleitung einer Behandlung mit Kortikosteroiden eine Verschlechterung oder keine Verbesserung eintritt, sollte die Kortikosteroiddosis auf 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent erhöht. Opdualag muss in diesem Fall dauerhaft abgesetzt werden.
Immunvermittelte Endokrinopathien
Schwere Endokrinopathien, einschliesslich Hypothyreose, Hyperthyreose, Nebenniereninsuffizienz (einschliesslich Nebennierenrindeninsuffizienz), Hypophysitis (einschliesslich Hypopituitarismus), Diabetes mellitus und diabetische Ketoazidose wurden bei der Behandlung mit Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ).
Die Patienten sollten auf klinische Anzeichen und Symptome von Endokrinopathien, sowie auf Hyperglykämie und Veränderungen der Schilddrüsenfunktion (zu Beginn der Behandlung, regelmässig während der Behandlung und wenn angezeigt nach klinischem Ermessen) überwacht werden. Bei den Patienten können Müdigkeit, Kopfschmerzen, Veränderungen des psychischen Zustandes, Bauchschmerzen, veränderte Stuhlgewohnheiten, Hypotonie oder unspezifische Symptome auftreten oder unspezifische Symptome, die anderen Ursachen wie Hirnmetastasen oder der zugrunde liegenden Erkrankung ähneln, beobachtet werden. Soweit keine alternative Ätiologie identifiziert wurde, sollten Anzeichen oder Symptome von Endokrinopathien als immunvermittelt betrachtet werden.
Bei einer symptomatischen Hypothyreose sollte Opdualag aufgeschoben und, soweit erforderlich, eine Schilddrüsenhormon-Substitutionstherapie eingeleitet werden. Bei einer symptomatischen Hyperthyreose sollte Opdualag aufgeschoben und, soweit erforderlich, eine Behandlung mit Thyreostatika eingeleitet werden. Kortikosteroide in einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent sollten in Betracht gezogen werden, wenn ein Verdacht auf eine akute Entzündung der Schilddrüse besteht. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids, falls erforderlich, wieder aufgenommen werden. Die Schilddrüsenfunktion sollte weiterhin überwacht werden, um sicherzustellen, dass eine angemessene Hormon-Substitutionstherapie verwendet wird. Bei lebensbedrohlicher (Grad 4) Hyperthyreose oder Hypothyreose muss Opdualag dauerhaft abgesetzt werden.
Bei einer symptomatischen Grad 2 Nebenniereninsuffizienz sollte Opdualag aufgeschoben und, soweit erforderlich, eine physiologische Kortikosteroid-Substitutionstherapie eingeleitet werden. Die Nebennierenfunktion und die Hormonspiegel sollten weiterhin überwacht werden, um sicherzustellen, dass eine angemessene Kortikosteroid-Substitutionstherapie angewendet wird.
Bei einer lebensbedrohlichen (Grad 4) Hypophysitis muss Opdualag dauerhaft abgesetzt werden. Bei symptomatischer Hypophysitis Grad 2 oder 3 sollte die Behandlung mit Opdualag aufgeschoben und bei Bedarf mit einer Hormonersatztherapie begonnen werden. Bei Verdacht auf akute Entzündung der Hypophyse sollte auch eine Behandlung mit Kortikosteroiden in einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent in Betracht gezogen werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids, falls erforderlich, wieder aufgenommen werden. Die Hypophysenfunktion und Hormonspiegel sollten weiterhin überwacht werden, um sicherzustellen, dass die passende Hormonersatztherapie angewandt wird.
Bei einem symptomatischen Diabetes sollte die Behandlung mit Opdualag aufgeschoben und bei Bedarf mit einer Insulinersatztherapie begonnen werden. Der Blutzuckerspiegel sollte überwacht werden, um sicherzustellen, dass eine passende Insulinersatztherapie angewandt wird. Bei einem lebensbedrohlichen Diabetes muss Opdualag dauerhaft abgesetzt werden.
Immunvermittelte Hautreaktionen
Schwerer Hautausschlag wurde unter Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ). Opdualag sollte bei Hautausschlag Grad 3 aufgeschoben und bei Hautausschlag Grad 4 abgesetzt werden. Schwerer Hautausschlag sollte mit hochdosierten Kortikosteroiden in einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalenten behandelt werden.
Seltene Fälle von Stevens-Johnson Syndrom (SJS) und toxischer epidermaler Nekrolyse (TEN; einschliesslich letaler Verläufe) wurden unter Nivolumab als Monotherapie beobachtet und könnten möglicherweise mit Nivolumab in Kombination mit Relatlimab auftreten. Wenn Symptome oder Anzeichen auf SJS oder TEN vermutet werden, sollte die Behandlung mit Opdualag aufgeschoben und der Patient an eine spezialisierte Abteilung zur Beurteilung und Behandlung überwiesen werden. Wird beim Patienten SJS oder TEN unter Verwendung von Opdualag bestätigt, so wird empfohlen Opdualag dauerhaft abzusetzen (siehe "Dosierung/Anwendung" ).
Vorsicht ist geboten, wenn für einen Patienten, der zuvor bei Behandlung mit anderen immunstimulierenden Krebsmedikamenten eine schwere oder lebensbedrohliche Hautreaktion erlitten hat, die Anwendung von Opdualag erwogen wird.
Immunvermittelte Myokarditis
Fälle von Myokarditis wurden unter Nivolumab in Kombination mit Relatlimab beobachtet (siehe "Unerwünschte Wirkungen" ). Die Diagnose von Myokarditis erfordert eine erhöhte Aufmerksamkeit. Patienten mit kardialen oder kardio-pulmonalen Symptomen sollten auf potenzielle Myokarditis untersucht werden. Falls ein Verdacht auf eine Myokarditis besteht, sollte unverzüglich eine Behandlung mit hochdosierten Kortikosteroiden (Prednison 1 bis 2 mg/kg/Tag oder Methylprednisolon 1 bis 2 mg/kg/Tag) und eine kardiologische Konsultation mit diagnostischer Abklärung gemäss aktuellen klinischen Richtlinien veranlasst werden. Wenn die Diagnose einer Myokarditis bestätigt wurde, sollte Opdualag, wie nachfolgend beschrieben, aufgeschoben oder dauerhaft abgesetzt werden.
Bei einer Myokarditis Grad 3 oder 4 muss Opdualag dauerhaft abgesetzt werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 2 bis 4 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden (siehe "Dosierung/Anwendung" ).
Bei einer Myokarditis Grad 2 sollte die Behandlung mit Opdualag aufgeschoben werden. Eine Behandlung mit Kortikosteroiden mit einer Dosis von 1 bis 2 mg/kg/Tag Methylprednisolon-Äquivalent sollte eingeleitet werden. Bei Erholung kann, nach Ausschleichen des Kortikosteroids, eine Wiederaufnahme der Behandlung mit Opdualag in Betracht gezogen werden. Wenn trotz der Einleitung einer Behandlung mit Kortikosteroiden eine Verschlechterung oder keine Verbesserung eintritt, sollte die Kortikosteroiddosis auf 2 bis 4 mg/kg/Tag Methylprednisolon-Äquivalent erhöht. Opdualag muss in diesem Fall dauerhaft abgesetzt werden (siehe "Dosierung/Anwendung" ).
Andere immunvermittelte unerwünschte Wirkungen
Die folgenden klinisch signifikanten immunvermittelten unerwünschten Wirkungen wurden selten bei Patienten unter Behandlung mit Nivolumab in Kombination mit Relatlimab berichtet: Uveitis, Pankreatitis, exokrine Pankreasinsuffizienz, Guillain-Barré-Syndrom, Myositis/Rhabdomyolyse, Myasthenia gravis, Enzephalitis, hämolytische Anämie, Vogt-Koyanagi-Harada-Syndrom (VKH), Zöliakie (siehe "Unerwünschte Wirkungen" ).
Die folgenden zusätzlichen klinisch signifikanten immunvermittelten unerwünschten Wirkungen wurden selten bei Patienten unter Behandlung mit Nivolumab als Monotherapie oder unter Behandlung mit Nivolumab in Kombination mit anderen genehmigten Therapeutika berichtet: Demyelinisierung, autoimmune Neuropathie (einschliesslich Fazialis- und Abduzensparese), Myasthenie-Syndrom, aseptische Meningitis, Gastritis, Sarkoidose, Duodenitis und Hypoparathyreoidismus.
Unter der Behandlung mit Immun-Checkpoint-lnhibitoren wurden Fälle von transverser Myelitis und aplastischer Anämie beobachtet. Die Patienten sollten hinsichtlich Anzeichen und Symptomen überwacht werden, die auf diese immunvermittelten Nebenwirkungen hindeuten.
Bei Verdacht auf immunvermittelte unerwünschte Wirkungen sollte eine angemessene Beurteilung erfolgen, um die Ätiologie zu bestätigen oder andere Ursachen auszuschliessen. Basierend auf der Schwere der unerwünschten Wirkungen sollte Opdualag aufgeschoben und Kortikosteroide verabreicht werden. Bei Erholung kann Opdualag nach Ausschleichen des Kortikosteroids wieder aufgenommen werden. Opdualag muss bei wiederauftretenden schweren immunvermittelten unerwünschten Wirkungen und allen lebensbedrohlichen immunvermittelten unerwünschten Wirkungen dauerhaft abgesetzt werden.
Patienten mit vorbestehender Autoimmunerkrankung
Bei Patienten mit vorbestehender Autoimmunerkrankung (AIE) deuten Daten aus Beobachtungsstudien auf ein erhöhtes Risiko für immunvermittelte unerwünschte Wirkungen nach Therapie mit Immun-Checkpoint-Inhibitoren im Vergleich zu Patienten ohne vorbestehende AIE hin. Darüber hinaus traten häufig Schübe der zugrundeliegenden AIE auf, die aber überwiegend leicht und gut behandelbar waren.
Andere wichtige Warnhinweise und Vorsichtsmassnahmen, inklusive Klasseneffekte
Bei mit PD-1-Inhibitoren behandelten Patienten wurde im Postmarketing-Umfeld Abstossungen von soliden Organtransplantaten beobachtet. Die Behandlung mit Nivolumab in Kombination mit Relatlimab kann das Abstossungsrisiko bei Empfängern solider Organtransplantate erhöhen. Bei diesen Patienten sollte der Nutzen der Behandlung mit Nivolumab in Kombination mit Relatlimab gegen das Risiko einer möglichen Organabstossung abgewogen werden.
Hämophagozytische Lymphohistiozytose (HLH) wurde bei Patienten unter Behandlung mit Nivolumab als Monotherapie, Nivolumab in Kombination mit Relatlimab und mit Nivolumab in Kombination mit anderen Therapeutika beobachtet. Unter Behandlung mit Nivolumab in Kombination mit Relatlimab wurde ein letaler Verlauf berichtet. Bei der Verabreichung von Nivolumab in Kombination mit Relatlimab ist Vorsicht geboten. Wenn HLH bestätigt wird, sollte die Anwendung von Nivolumab in Kombination mit Relatlimab abgebrochen und eine Behandlung der HLH eingeleitet werden.
Fälle schnell einsetzender und schwerwiegender graft-versus-host-disease (GVHD), einschliesslich letaler Verläufe, wurde bei Patienten berichtet, welche vor oder nach einer allogenen hämatopoetischen Stammzell-Transplantation (HSCT) mit Nivolumab behandelt wurden. Die Behandlung mit Nivolumab in Kombination mit Relatlimab kann das Risiko einer schweren GVHD und auf Tod bei Patienten mit vorgängiger HSCT erhöhen, vor allem bei Patienten mit GVHD in der Vorgeschichte. Bei diesen Patienten sollte der Nutzen einer Behandlung mit Nivolumab in Kombination mit Relatlimab gegenüber dem möglichen Risiko abgewogen werden.
Infusionsreaktionen
In klinischen Studien mit Nivolumab in Kombination mit Relatlimab wurde über schwere Infusionsreaktionen berichtet (siehe "Unerwünschte Wirkungen" ). Im Fall einer schweren oder lebensbedrohlichen Infusionsreaktion muss die Opdualag-Infusion abgebrochen werden und eine geeignete medizinische Behandlung erfolgen. Patienten mit einer leichten oder mittelschweren Infusionsreaktion können Opdualag erhalten, vorausgesetzt sie werden engmaschig überwacht und erhalten eine Prämedikation gemäss lokalen Richtlinien zur Vorbeugung von Infusionsreaktionen.
Patienten, welche von der pivotalen Studie zum nicht resezierbaren oder metastasierten Melanom ausgeschlossen wurden
Patienten mit aktiver Autoimmunerkrankung, Erkrankungen, die eine systemische Behandlung mit mässig- oder hochdosierten Kortikosteroiden oder immunsuppressiven Medikamenten erfordern, sowie aktive oder unbehandelte Hirnmetastasen oder leptomeningeale Metastasen wurden von der Studie ausgeschlossen. Da keine Daten vorliegen, sollte Nivolumab in Kombination mit Relatlimab bei diesen Patientenpopulationen mit Vorsicht, d.h. nach sorgfältiger individueller Nutzen-Risiko-Abwägung mit angewendet werden.
Interaktionen
Nivolumab und Relatlimab sind humane monoklonale Antikörper, darum wurden keine Interaktionsstudien durchgeführt. Da monoklonale Antikörper nicht durch Cytochrom-P450-(CYP)-Enzyme oder andere Arzneimittel metabolisierende Enzyme metabolisiert werden, wird nicht erwartet, dass die Hemmung oder Induktion dieser Enzyme durch gleichzeitig verabreichte Arzneimittel die Pharmakokinetik von Nivolumab oder Relatlimab beeinträchtigt.
Pharmakodynamische Interaktionen
Systemische Immunsuppression
Die Verwendung von systemischen Kortikosteroiden und anderen Immunsuppressiva vor Beginn (Baseline) der Behandlung mit Nivolumab in Kombination mit Relatlimab sollte wegen ihrer möglichen Interferenz mit der pharmakodynamischen Aktivität vermieden werden. Systemische Kortikosteroide und andere Immunsuppressiva können jedoch nach Beginn der Therapie mit Nivolumab in Kombination mit Relatlimab zur Behandlung von immunvermittelten unerwünschten Wirkungen verwendet werden.
Schwangerschaft, Stillzeit
Frauen im gebärfähigen Alter/Verhütung
Bei gebärfähigen Frauen, die keine wirksame Empfängnisverhütung verwenden, ist Opdualag nicht empfohlen, es sei denn, der klinische Nutzen überwiegt das mögliche Risiko. Eine wirksame Empfängnisverhütung sollte für mindestens 5 Monate nach der letzten Gabe von Opdualag angewendet werden.
Schwangerschaft
Zur Anwendung von Nivolumab in Kombination mit Relatlimab bei schwangeren Frauen liegen keine Daten vor. Basierend auf dem Wirkmechanismus und auf Tierstudien kann Nivolumab in Kombination mit Relatlimab, einer schwangeren Frau verabreicht, den Fötus schädigen. Tierstudien haben eine embryofetale Toxizität mit Nivolumab gezeigt (siehe "Präklinische Daten" ). Es ist bekannt, dass humanes IgG4 die Plazentaschranke überwindet. Nivolumab und Relatlimab sind ein IgG4. Daher besteht die Möglichkeit, dass Nivolumab und Relatlimab von der Mutter auf den sich entwickelnden Fötus übergehen. Opdualag ist während einer Schwangerschaft nicht empfohlen, es sei denn, dass eine Behandlung mit Nivolumab/Relatlimab aufgrund des klinischen Zustandes der Frau erforderlich ist.
Stillzeit
Es ist nicht bekannt, ob Nivolumab und/oder Relatlimab in die menschliche Muttermilch übergehen. Da viele Medikamente, darunter Antikörper, in die Muttermilch sezerniert werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Der Nutzen des Stillens für das Kind und der Nutzen der Behandlung mit Opdualag für die Mutter sind gegeneinander abzuwägen und es muss entschieden werden, ob das Stillen oder die Behandlung beendet wird.
Fertilität
Es wurden keine Studien zur Beurteilung der Wirkung von Nivolumab und/oder Relatlimab auf die Fruchtbarkeit durchgeführt. Daher ist die Wirkung von Nivolumab und/oder Relatlimab auf die männliche und weibliche Fruchtbarkeit unbekannt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Opdualag kann einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben. Wegen möglicher unerwünschter Wirkungen wie Müdigkeit (siehe "Unerwünschte Wirkungen" ) sollte den Patienten geraten werden, beim Führen von Fahrzeugen oder Bedienen von Maschinen Vorsicht walten zu lassen, bis sie sicher sind, dass Opdualag keine nachteiligen Auswirkungen auf sie hat.
Die Patienten sollten darauf hingewiesen werden, nicht Auto zu fahren oder Maschinen zu bedienen, wenn sie Müdigkeit oder Schwindelgefühl verspüren.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Nivolumab in Kombination mit Relatlimab wurde bei 722 Patienten mit nicht resezierbarem oder metastasiertem Melanom (Studie CA224047 und CA224020) untersucht.
Die Häufigkeiten in der nachfolgenden Tabelle 2 basieren auf berichteten unerwünschten Arzneimittelwirkungen, unabhängig von der Beurteilung der Kausalität durch den Prüfarzt. Bei einer minimalen Nachbeobachtungszeit von 0,03 Monaten und einer medianen Nachbeobachtungszeit von 20,34 Monaten, waren die häufigsten unerwünschten Wirkungen (≥10%) Ermüdung (41%), Schmerzen des Muskel- und Skelettsystems (30%), Hautausschlag (23%), Pruritus (22%), Arthralgie (22%), Übelkeit (22%), Diarrhö (22%), Kopfschmerzen (17%), verminderter Appetit (14%), Husten (14%), Abdominalschmerz (14%), Verstopfung (13%), Hypothyreose (13%), Pyrexie (12%), Dyspnoe (10%) und Erbrechen (10%). Die häufigsten schwerwiegenden unerwünschten Wirkungen (≥1%) waren Anämie (1,1%), Pyrexie (1,1%), Kolitis (1,1%), Abdominalschmerz (1,0%), Diarrhö (1,0%) und Dyspnoe (1,0%). Unerwünschte Wirkungen von Grad 3-5, bei Patienten mit nicht resezierbarem oder metastasiertem Melanom, traten bei 42% der Patienten unter Nivolumab in Kombination mit Relatlimab und bei 38% der Patienten, die mit Nivolumab behandelt wurden auf.
Liste der unerwünschten Wirkungen
Die im Datensatz für Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, berichteten unerwünschten Wirkungen sind in Tabelle 2 dargestellt. Diese Wirkungen sind nach Systemorganklasse und Häufigkeit geordnet aufgeführt. Die Häufigkeiten sind folgendermassen definiert: sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10); gelegentlich (≥1/1'000 bis < 1/100); selten (≥1/10'000 bis < 1/1'000); sehr selten (< 1/10'000), nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden). Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 2: Unerwünschte
Wirkungen in klinischen
Studien – alle Grade
Infektionen und parasitäre
Erkrankungen
Häufig Infektion der oberen Atemwege, Follikulitis
Gelegentlich Enzephalitis, C-reaktives Protein erhöht, Blutkörperchensenkung
erhöht
Erkrankungen des Blutes und
des Lymphsystems
Sehr häufig Anämie (44,2%)a, Lymphopenie (38,9%)a, Leukopenie (13,6%)a,
Neutropenie (12,9%)a
Häufig Thrombozytopeniea, Eosinophilie, Laktatdehydrogenase im Blut erhöht
Gelegentlich Hämolytische Anaemie
Endokrine Erkrankungen
Sehr häufig Hypothyreose (12,5%)
Häufig Nebenniereninsuffizienz, Hyperthyreose, Thyroiditis
Gelegentlich Hypophysitis, Hypopituitarismus, Hypogonadismus
Stoffwechsel- und
Ernährungsstörungen
Sehr häufig Hyponatriämie (26,0%)a, Hyperkaliämie (17,0%)a, Hypokalzämie
(17,6%)a, verminderter Appetit (14,0%), Hypomagnesiämie (12,2%)a
Häufig Diabetes mellitus, Gewicht erniedrigt, Hyperkalzämiea,
Hypokaliämiea, Hypermagnesiämiea, Hypernatriämiea, Hypoglykämiea,
Hypoalbuminämie, Dehydratation, Hyperurikämie
Psychiatrische Erkrankungen
Häufig Verwirrtheitszustand
Erkrankungen des Nervensyste
ms
Sehr häufig Kopfschmerzen (17,2%)
Häufig Schwindelgefühl, periphere Neuropathie, Dysgeusie
Gelegentlich Guillain-Barré-Syndrom, Optikusneuritis, transverse Myelitis,
Myasthenia gravis
Augenerkrankungen
Häufig Uveitis, Sehverschlechterung, trockenes Auge
Gelegentlich Vogt-Koyanagi-Harada Krankheit, Tränensekretion verstärkt, okuläre
Hyperämie
Herzerkrankungen
Häufig Troponin erhöht
Gelegentlich Myokarditis, Perikarderguss
Gefässerkrankungen
Gelegentlich Phlebitis
Erkrankungen der Atemwege,
des Brustraums und
Mediastinums
Sehr häufig Husten (14,3%), Dyspnoe (10,5%)
Häufig Pneumonitisb, Nasenverstopfung
Gelegentlich Asthma, Pleuraerguss
Erkrankungen des Gastrointes
tinaltrakts
Sehr häufig Übelkeit (21,5%), Diarrhö (22,0%), Abdominalschmerz (14,4%),
Verstopfung (12,7%), Erbrechen (10,4%)
Häufig Pankreatitis, Kolitis, Gastritis, trockener Mund, Lipase erhöht,
Amylase erhöht, Stomatitis, Dysphagie
Gelegentlich Ösophagitis
Selten Exokrine Pankreasinsuffizienz
Nicht bekannt Zöliakie
Leber- und Gallenerkrankunge
n
Sehr häufig AST erhöht (29,2%)a, ALT erhöht (25,0%)a, alkalische Phosphatase
erhöht (22,3%)a
Häufig Hepatitis, Bilirubin erhöhta, Gamma-Glutamyltransferase erhöht
Gelegentlich Cholangitis
Erkrankungen der Haut und
des Unterhautgewebes
Sehr häufig Hautausschlag (23,4%), Pruritus (22,0%)
Häufig Vitiligo, Alopezie, trockene Haut, Urtikaria
Gelegentlich Pemphigoid, lichenoide Keratose, Lichtempfindlichkeitsreaktion,
Psoriasis
Skelettmuskulatur-,
Bindegewebs- und Knochenerkr
ankungen
Sehr häufig Schmerzen des Muskel- und Skelettsystems (29,9%), Arthralgie (21,9%)
Häufig Arthritis, muskuläre Schwäche, Muskelspasmen
Gelegentlich Myositis, systemischer Lupus erythematodes, Polymyalgia rheumatica,
Sjögren Syndrom, rheumatoide Arthritis, Bursitis
Erkrankungen der Nieren und
Harnwege
Sehr häufig Kreatinin erhöht (20,3%)a
Häufig Nierenversagen, Proteinurie
Gelegentlich Nephritis
Erkrankungen der Geschlechts
organe und der Brustdrüse
Gelegentlich Azoospermie
Allgemeine Erkrankungen und
Beschwerden am Verabreichung
sort
Sehr häufig Ermüdung (41,1%), Pyrexie (11,5%)
Häufig Ödem, grippeähnliche Erkrankung, Schüttelfrost
Selten Serositis
Verletzung, Vergiftung und
durch Eingriffe bedingte
Komplikationen
Häufig Reaktion im Zusammenhang mit einer Infusion
a Häufigkeiten von Laborbegriffen spiegeln den Anteil der Patienten wider, bei denen eine Verschlechterung der Labormesswerte gegenüber dem Ausgangswert auftrat.
b Fall mit tödlichem Verlauf wurde in der klinischen Studie CA224047 berichtet.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Nivolumab und/oder Relatlimab sind mit immunvermittelten unerwünschten Wirkungen assoziiert. Die meisten immunvermittelten unerwünschten Wirkungen konnten durch entsprechende medizinische Behandlung behoben werden. Die Behandlungsrichtlinien für diese unerwünschten Wirkungen sind in "Warnhinweise und Vorsichtsmassnahmen" beschrieben.
Immunvermittelte Pneumonitis
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Pneumonitis, darunter interstitielle Lungenerkrankung und Lungeninfiltrat, 3,7% (27/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 0,6% (4/722). Letale Ereignisse traten bei 0,3% (2/722) der Patienten auf. Die mediane Zeit bis zum Auftreten betrug 28,1 Wochen (Bereich: 3,6-170,0). Bei 21/27 Patienten (77,8%) gingen die Symptome nach einer medianen Zeit von 10 Wochen (Bereich: 0,6-133,0+) vollständig zurück. Eine immunvermittelte Pneumonitis führte bei 1,0% der Patienten zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und erforderte bei 55,6% der Patienten mit immunvermittelter Pneumonitis hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Immunvermittelte Kolitis
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Diarrhö, Kolitis oder häufiger Stuhlgang 23,7% (171/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 3,5% (25/722). Die mediane Zeit bis zum Auftreten betrug 13,4 Wochen (Bereich: 0,1-271,3). Bei 152/171 Patienten (88,9%) gingen die Symptome nach einer medianen Zeit von 2,3 Wochen (Bereich: 0,1-289,1+) vollständig zurück. Eine immunvermittelte Kolitis führte bei 1,7% der Patienten zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und erforderte bei 21,1% der Patienten mit immunvermittelter Kolitis hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Immunvermittelte Hepatitis
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Veränderungen der Leberwerte 19,0% (137/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 5,1% (37/722). Die mediane Zeit bis zum Auftreten betrug 8,1 Wochen (Bereich: 0,1-198,0). Bei 95/134 Patienten (70,9%) gingen die Symptome nach einer medianen Zeit von 9,1 Wochen (Bereich: 0,6-280,6+) vollständig zurück. Bei 1,8% der Patienten kam es zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und 24,1% der Patienten benötigten hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Immunvermittelte Nephritis und Nierenfunktionsstörung
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Nephritis oder Nierenfunktionsstörung 7,5% (54/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 1,2% (9/722). Die mediane Zeit bis zum Auftreten betrug 20,4 Wochen (Bereich: 0,1-210,4). Bei 39/52 Patienten (75,0%) gingen die Symptome nach einer medianen Zeit von 8,0 Wochen (Bereich: 0,1-274,1+) vollständig zurück. Eine immunvermittelte Nephritis oder Nierenfunktionsstörung führte bei 0,7% der Patienten zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und erforderte bei 7,5% der Patienten mit immunvermittelter Nephritis oder Nierenfunktionsstörung hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Immunvermittelte Endokrinopathien
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Schilddrüsenfunktionsstörung, inklusive Hypothyreose oder Hyperthyreose, 16,3% (118/722). Es gab keine Fälle von Schilddrüsenfunktionsstörung Grad 3/4. Die Inzidenz von Grad 3/4 Nebenniereninsuffizienz Ereignissen betrug 1,1% (8/722). Die Inzidenz von Grad 3/4 Hypophysitis Ereignissen betrug 0,6% (4/722). Es gab keine Fälle von Grad 3/4 Hypopituitarismus. Die Inzidenz von Grad 3/4 Diabetes mellitus (inklusive Typ 1 Diabetes mellitus) Ereignissen betrug 0,4% (3/722). Die mediane Zeit bis zum Auftreten betrug 15,9 Wochen (Bereich: 1,0-223,7). Bei 39/151 Patienten (25,8%) gingen die Symptome innerhalb eines Zeitraums zwischen 0,4-328,0+ Wochen vollständig zurück. Immunvermittelte Endokrinopathien führten bei 0,6% der Patienten zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und erforderten bei 12,6% der Patienten mit immunvermittelten Endokrinopathien hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Immunvermittelte Hautreaktionen
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Hautausschlag 41,4% (299/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 1,4% (10/722). Die mediane Zeit bis zum Auftreten betrug 5,9 Wochen (Bereich: 0,1-142,1). Bei 144/297 Patienten (48,5%) gingen die Symptome nach einer medianen Zeit von 93,4 Wochen (Bereich: 0,1-318,0+) vollständig zurück. Immunvermittelte Hautreaktionen führten bei 0,4% der Patienten zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und erforderten bei 4,3% der Patienten mit immunvermittelten Hautreaktionen hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Immunvermittelte Myokarditis
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Myokarditis 0,8% (6/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 0,4% (3/722). Die mediane Zeit bis zum Auftreten betrug 5,1 Wochen (Bereich: 2,1-8,1). Bei 6/6 Patienten (100%) gingen die Symptome nach einer medianen Zeit von 3 Wochen (Bereich: 0,6-14,0) vollständig zurück. Eine immunvermittelte Myokarditis führte bei 0,8% der Patienten zum dauerhaften Abbruch von Nivolumab in Kombination mit Relatlimab und erforderte bei 100% der Patienten mit immunvermittelter Myokarditis hochdosierte Kortikosteroide (Prednison ≥40 mg pro Tag oder äquivalent).
Infusionsreaktionen
Bei Patienten, welche mit Nivolumab in Kombination mit Relatlimab behandelt wurden, betrug die Inzidenz von Überempfindlichkeits-/Infusionsreaktionen 4,8% (35/722). Die Inzidenz von Grad 3/4 Ereignissen betrug 0,3% (2/722).
Immunogenität
Die Inzidenz von Anti-Drug-Antikörpern (ADAs) hängt in hohem Mass von der Empfindlichkeit und Spezifität des Tests ab. Darüber hinaus kann die beobachtete Inzidenz der Antikörperpositivität (einschliesslich neutralisierender Antikörper) in einem Test von mehreren Faktoren beeinflusst werden, beispielsweise von der Testmethodik, der Probenhandhabung, dem Zeitpunkt der Probengewinnung, den Begleitmedikamenten und der Grundkrankheit. Aus diesem Grund kann der Vergleich der Inzidenz von Antikörpern gegen Nivolumab oder Relatlimab mit der Inzidenz von Antikörpern gegen andere Arzneimittel irreführend sein.
Bei Patienten der Studien CA224047 und CA224020, welche mit Nivolumab in Kombination behandelt wurden, lag die Inzidenz von behandlungsbedingten Anti-Relatlimab-Antikörpern und neutralisierenden Antikörpern gegen Relatlimab bei 4,5% (26/577) bzw. 0,2% (1/577) der auswertbaren Patienten für Anti-Medikamenten-Antikörper. Die Inzidenz von behandlungsbedingten Anti-Nivolumab-Antikörpern und neutralisierenden Antikörpern gegen Nivolumab betrug 3,4% (20/588) bzw. 0,3% (2/588) und war damit ähnlich hoch wie die in der Nivolumab-Gruppe beobachtete Inzidenz von 5,9% (16/272) bzw. 0,4% (1/272).
Pädiatrische Population
Die Sicherheit von Opdualag bei nicht resezierbarem oder metastasiertem Melanom wurde bei Kindern und Jugendlichen nicht nachgewiesen (siehe "Dosierung/Anwendung" ).
Ältere Patienten
Von den 355 Patienten, die mit Opdualag behandelt wurden, waren 47% ≥65 Jahre, 29% waren 65 bis 74 Jahre, 17% waren 75 bis 84 Jahre, 19% waren ≥75 Jahre und 2% waren ≥85 Jahre alt. Insgesamt wurden keine Unterschiede in der Sicherheit zwischen älteren (≥65 Jahre) und jüngeren Patienten berichtet (siehe "Eigenschaften/Wirkungen" ).
Meldung des Verdachts auf unerwünschten Wirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle einer Überdosierung berichtet.
Eigenschaften/Wirkungen
ATC-Code
L01FY02
Pharmakotherapeutische Gruppe: Antineoplastische Wirkstoffe, Kombinationen von monoklonalen Antikörpern und Antikörper-Wirkstoff-Konjugaten
Wirkungsmechanismus
Opdualag ist eine Fixdosis-Kombination aus Nivolumab (Anti-PD-1) und Relatlimab (Anti-LAG-3).
LAG-3 ist ein T-Zell-Rezeptor, der an seine Liganden, wie den Haupthistokompatibilitätskomplex Klasse II, bindet und einen hemmenden Immunsignalweg reguliert, der die T-Zell-Proliferation, die Effektor-Funktion, die Zytokin-Produktion und die Entwicklung von Gedächtnis-T-Zellen hemmt. Relatlimab ist ein humaner monoklonaler IgG4-Antikörper, der an den LAG-3-Rezeptor bindet und die durch LAG-3 vermittelte Hemmung der Immunantwort aufhebt, indem er seine Interaktion mit Liganden blockiert. Relatlimab zeigt in vitro eine funktionelle Aktivität bei der Wiederherstellung der Effektor-Funktion von T-Zellen und der Förderung der Zytokin-Signalisierung.
Die Bindung der PD-1-Liganden PD-L1 und PD-L2 an den PD-1-Rezeptor, der sich auf T-Zellen befindet, hemmt die T-Zell-Proliferation und die Zytokin-Produktion. Eine Hochregulierung der PD-1-Liganden tritt bei einigen Tumoren auf, und die Signalübertragung über diesen Weg kann zur Hemmung der aktiven T-Zell-Immunüberwachung von Tumoren beitragen. Nivolumab ist ein humaner monoklonaler IgG4-Antikörper, der an den PD-1-Rezeptor bindet und dessen Interaktion mit PD-L1 und PD-L2 blockiert, wodurch die durch den PD-1-Signalweg vermittelte Hemmung der Immunantwort, einschliesslich der Anti-Tumor-Immunantwort, aufgehoben wird. In syngenen Mäusetumormodellen führte die Blockierung der PD-1-Aktivität zu einem verringerten Tumorwachstum.
LAG-3 und PD-1, zwei eigenständige inhibitorische Immun-Checkpoint-Signalwege, wirken synergistisch auf Effektor-T-Zellen, was zur Entwicklung einer T-Zell-Erschöpfung und beeinträchtigten zytotoxischen Funktion führt. Die kombinierte Hemmung durch Relatlimab (Anti-LAG-3) und Nivolumab (Anti-PD-1) ermöglicht eine T-Zell-Aktivierung und stellt die Effektor-Funktion der T-Zellen wieder her. Die Wirkung der kombinierten Hemmung übersteigt dabei die Wirkung jedes einzelnen Antikörpers allein. In syngenen Tumormodellen der Maus potenziert die LAG-3-Blockade die Anti-Tumor-Aktivität der PD-1-Blockade, hemmt das Tumorwachstum und fördert die Tumorregression.
Pharmakodynamik
Pharmakodynamische Veränderungen von Interferon γ (IFNγ)
In der Studie CA224047 stieg der IFNγ-Spiegel im Blut nach der Verabreichung von Relatlimab in Kombination mit Nivolumab oder Nivolumab allein an. Der Anstieg des IFNγ-Spiegels war im Nivolumab in Kombination mit Relatlimab-Arm grösser (medianer Anstieg von 105,6% und 108,0% im Talspiegel nach der ersten bzw. zweiten Dosis) als im Nivolumab-Arm (medianer Anstieg von 56,3% und 50,7% im Talspiegel nach der ersten bzw. zweiten Dosis). Diese Daten sprechen für eine erhöhte T-Zell-Aktivierung und IFNγ-Produktion durch Nivolumab in Kombination mit Relatlimab.
Klinische Wirksamkeit
In diesem Abschnitt werden die klinischen Erfahrungen mit Opdualag, Fixdosis-Kombination aus Nivolumab und Relatlimab, bei Patienten mit nicht resezierbarem oder metastasiertem Melanom vorgestellt.
Randomisierte Phase-2/3-Studie von Nivolumab in Kombination mit Relatlimab vs. Nivolumab (CA224047)
Die Sicherheit und Wirksamkeit von Nivolumab in Kombination mit Relatlimab zur Behandlung von zuvor unbehandeltem metastasiertem oder nicht resezierbarem Melanom wurde in einer randomisierten, doppelt verblindeten Phase-2/3-Studie untersucht (CA224047). In die Studie wurden Patienten mit einem ECOG-Performance-Status-Score von 0 oder 1 und einem histologisch bestätigten Melanom im Stadium III (nicht resezierbar) oder im Stadium IV gemäss American Joint Committee on Cancer (AJCC) Version 8 aufgenommen. Die Patienten durften eine vorherige adjuvante oder neoadjuvante Melanomtherapie erhalten haben (Anti-PD-1-, Anti-CTLA-4- oder BRAF-MEK-Therapie war erlaubt, solange zwischen der letzten Dosis der Therapie und dem Datum des Rezidivs mindestens 6 Monate lagen; eine Interferontherapie war erlaubt, solange die letzte Dosis mindestens 6 Wochen vor der Randomisierung lag). Patienten mit aktiver Autoimmunerkrankung, Erkrankungen, die eine systemische Behandlung mit mässig- oder hochdosierten Kortikosteroiden oder immunsuppressiven Medikamenten erfordern, Aderhautmelanom und Patienten mit aktiven oder unbehandelten Hirn- oder leptomeningealen Metastasen waren von der Studie ausgeschlossen.
Insgesamt 714 Patienten wurden randomisiert und erhielten entweder Nivolumab in Kombination mit Relatlimab (n = 355) oder Nivolumab (n = 359). Die Patienten im Kombinationsarm erhielten 480 mg Nivolumab/160 mg Relatlimab über 60 Minuten alle 4 Wochen. Die Patienten im Nivolumab-Arm erhielten 480 mg Nivolumab alle 4 Wochen. Die Randomisierung erfolgte stratifiziert nach Tumor-PD-L1 (≥1% vs. < 1%) unter Verwendung des PD-L1 IHC 28-8 pharmDx-Tests und der LAG-3-Expression (≥1% vs. < 1%), die mit einem analytisch validierten LAG-3 IHC-Assay bestimmt wurde, dem BRAF V600-Mutationsstatus und dem M-Stadium gemäss dem AJCC Version 8 Staging-System (M0/M1any[0] vs. M1any[1]). Die Patienten wurden bis zur Krankheitsprogression oder inakzeptabler Toxizität behandelt. Tumorbeurteilungen gemäss den Response Evaluation Criteria in Solid Tumours (RECIST), Version 1.1, wurden 12 Wochen nach der Randomisierung durchgeführt und alle 8 Wochen bis zu 52 Wochen fortgesetzt, danach alle 12 Wochen bis zum Fortschreiten der Erkrankung oder zum Abbruch der Behandlung, je nachdem, was später eintrat. Der primäre Wirksamkeitsendpunkt war das progressionsfreie Überleben (PFS) in der Zielpopulation (ITT), das durch eine verblindete unabhängige zentrale Überprüfung (BICR) bestimmt wurde. Das Gesamtüberleben (OS) in der ITT-Population war ein sekundärer Endpunkt.
Die Ausgangscharakteristika in der ITT-Population waren zwischen den beiden Gruppen ausgeglichen. Das mediane Alter betrug 63 Jahre (Bereich: 20-94), 58% waren Männer und 97% waren weiss. Der ECOG-Performance-Status zu Beginn der Studie war 0 (67%) oder 1 (33%). Die Mehrheit der Patienten hatte eine Erkrankung im AJCC-Stadium IV (92%); 38,9% hatten M1c, 2,4% M1d, 36% hatten bei Studienbeginn einen LDH-Ausgangswert grösser als ULN. 39% der Patienten hatten ein BRAF-Mutation-positives Melanom, 75% hatten LAG-3 ≥1% und 41% der Patienten hatten PD-L1 ≥1% Tumorzellmembran-Expression. Bei den Patienten mit quantifizierbarer Tumor-PD-L1-Expression war die Verteilung der Patienten auf die beiden Behandlungsgruppen ausgeglichen.
Bei der Primäranalyse in der ITT-Population mit einer medianen Nachbeobachtungszeit von 13,2 Monaten (Spanne: 0-33,1 Monate) wurde eine statistisch signifikante Verbesserung des PFS beobachtet, mit einem medianen PFS von 10,1 Monaten in der Gruppe von Patienten, die mit Nivolumab in Kombination mit Relatlimab behandelt wurden, im Vergleich zu 4,6 Monaten in der Nivolumab-Gruppe (HR = 0,75; 95% KI: 0,62, 0,92; p = 0,0055). Zum Zeitpunkt der präspezifizierten finalen OS-Analyse in der ITT-Population mit einer medianen Nachbeobachtungszeit von 19,3 Monaten war das OS nicht statistisch signifikant (HR = 0,80; 95% KI: 0,64, 1,01).
In einer nachfolgenden deskriptiven Analyse mit einer medianen Nachbeobachtungszeit von 34,9 Monaten betrug das mediane PFS 10,2 Monate (95% KI: 6,51, 15,74) in der Nivolumab + Relatlimab-Gruppe und 4,6 Monate (95% KI: 3,48, 6,47) in der Nivolumab-Gruppe (HR = 0,78; 95% KI: 0,65, 0,93). Das mediane OS betrug 53,3 Monate (95% KI: 34,04, NR) in der Nivolumab + Relatlimab-Gruppe und 33,2 Monate (95% KI: 25,23, 45,77) in der Nivolumab-Gruppe (HR = 0,77; 95% KI: 0,64, 0,94).
Es erfolgten deskriptive OS-Subgruppenanalysen nach PD-L1-Expression mit Cut-off 1% und 10%, welche im Anschluss dargestellt sind; die Studie war nicht darauf ausgelegt, statistische Unterschiede zwischen den Behandlungen innerhalb der verschiedenen PD-L1-Cut-off-Bereiche nachzuweisen.
Abbildung 1: Kaplan-Meier-Kurve für das OS bei einer PD-L1-Expression < 1% zu Studienbeginn
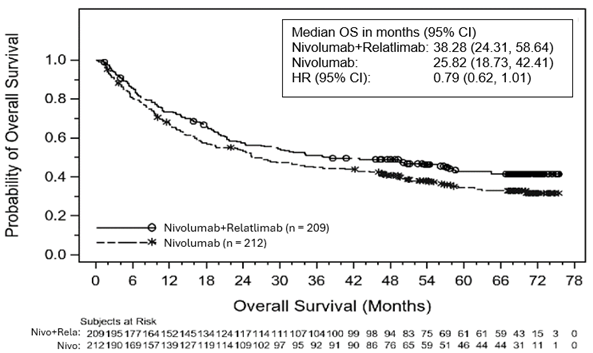
Abbildung 2: Kaplan-Meier-Kurve für das OS bei einer PD-L1-Expression ≥1% zu Studienbeginn
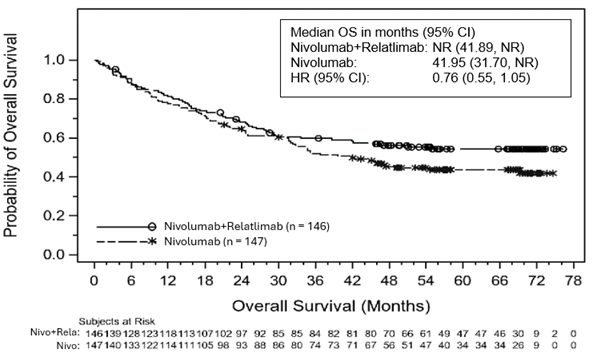
Abbildung 3: Kaplan-Meier-Kurve für das OS bei einer PD-L1-Expression < 10% zu Studienbeginn
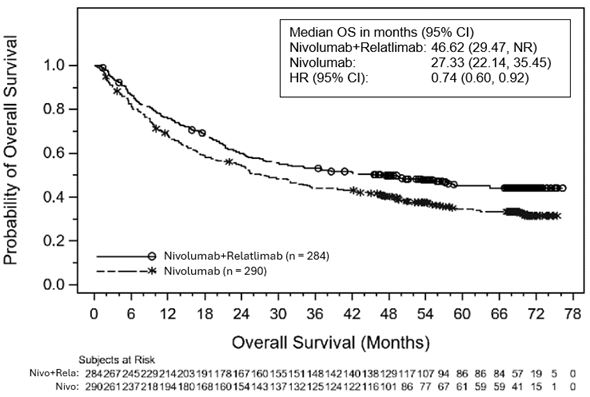
Abbildung 4: Kaplan-Meier-Kurve für das OS bei einer PD-L1-Expression ≥10% zu Studienbeginn
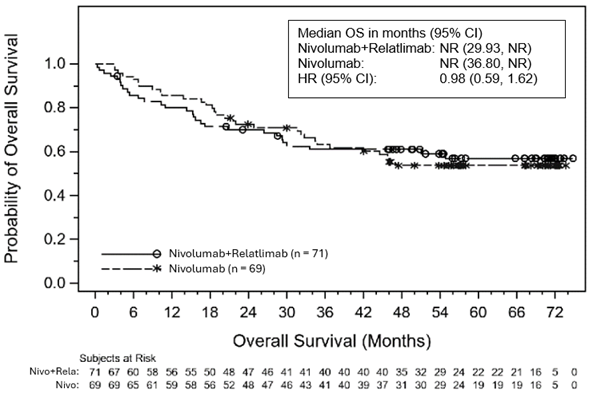
Pharmakokinetik
Die Pharmakokinetik (PK) von Nivolumab und Relatlimab nach Verabreichung von Nivolumab in Kombination mit Relatlimab wurde bei Patienten mit verschiedenen Krebsarten charakterisiert. Die Patienten erhielten Relatlimab-Dosen von 20 bis 800 mg alle 2 Wochen und 160 bis 1440 mg alle 4 Wochen entweder als Monotherapie oder in Kombination mit Nivolumab-Dosen von 80 oder 240 mg alle 2 Wochen oder 480 mg alle 4 Wochen.
Steady-State-Konzentrationen von Relatlimab wurden bei einer Verabreichung alle 4 Wochen nach 16 Wochen erreicht, und die systemische Akkumulation betrug das 1,9-fache. Die durchschnittliche Konzentration (Cavg) von Relatlimab nach der ersten Dosis stieg bei Dosen ≥160 mg alle 4 Wochen dosisproportional an.
Nach der Kombination von 480 mg Nivolumab und 160 mg Relatlimab in fixer Dosierung alle 4 Wochen betrugen die geometrischen Mittelwerte (CV%) der maximalen Konzentration (Cmax), der minimalen Konzentration (Cmin) und der Cavg von Relatlimab im Steady-State 62,2 μg/ml (30,1%), 15,3 μg/ml (64,3%) bzw. 28,8 μg/ml (44,8%). Der geometrische Mittelwert (CV%) der Cmax, der Cmin und der Cavg von Nivolumab im Steady-State betrugen 187 μg/ml (32,9%), 59,7 μg/ml (58,6%) bzw. 94,4 μg/ml (43,3%).
Der geometrische Mittelwert der Nivolumab Cmin mit 0,931 (95% KI: 0,855-1,013) im Steady-State war bei kombinierter Verabreichung von Relatlimab und Nivolumab ähnlich wie bei Verabreichung von Nivolumab alleine.
Absorption
Nicht zutreffend.
Distribution
Der geometrische Mittelwert (CV%) für das Verteilungsvolumen von Relatlimab im Steady-State liegt bei 6,65 l (19,8%) und für Nivolumab bei 6,65 l (19,2%).
Metabolismus
Der Metabolismus von Nivolumab und Relatlimab wurde nicht ermittelt. Als vollständige humane monoklonale IgG4-Antikörper wird erwartet, dass Nivolumab und Relatlimab ebenso wie endogenes IgG via kleine Peptide und Aminosäuren katabolisch abgebaut werden.
Elimination
Die Clearance (CL) von Relatlimab ist im Steady-State um 9,7% niedriger [geometrisches Mittel (CV%), 5,48 ml/h (41,3%)] als nach der ersten Dosis [6,06 ml/h (38,9%)]. Nach Verabreichung von Nivolumab 480 mg und Relatlimab 160 mg im Abstand von 4 Wochen beträgt die mittlere (CV%) effektive Halbwertszeit von Relatlimab 26,2 Tage (37%).
Die Nivolumab-Clearance ist im Steady-State um 21,1% niedriger [geometrisches Mittel (CV%), 7,57 ml/h (40,1%)] als nach der ersten Dosis [9,59 ml/h (40,3%)] und die mittlere (CV%) terminale Halbwertszeit (t1/2) beträgt 26,5 Tage (36,4%).
Kinetik spezieller Patientengruppen
Basierend auf einer Populations-PK-Analyse zeigte sich für die folgenden Faktoren kein klinisch bedeutsamer Einfluss auf die CL von Nivolumab und Relatlimab: Alter (Bereich: 17 bis 92 Jahre), Geschlecht oder Rasse.
Leberfunktionsstörungen
Die Auswirkung von Leberfunktionsstörungen auf die Clearance von Nivolumab und Relatlimab wurde mittels Populations-PK-Analyse bei Patienten mit leichter oder mässiger Leberfunktionsstörung (definiert nach den Kriterien des US National Cancer Institute für Leberfunktionsstörungen) im Vergleich zu Patienten mit normaler Leberfunktion untersucht. Es wurden keine klinisch bedeutsamen Unterschiede in der Clearance von Nivolumab oder Relatlimab zwischen Patienten mit leichten und moderaten Leberfunktionsstörungen und Patienten mit normaler Leberfunktion festgestellt. Für Patienten mit stark eingeschränkter Leberfunktion liegen nicht genügend Daten vor, um Schlussfolgerungen für diese Patientenpopulation ziehen zu können.
Nierenfunktionsstörungen
Die Auswirkung von Nierenfunktionsstörungen auf die Clearance von Nivolumab und Relatlimab wurde durch eine Populations-PK-Analyse bei Patienten mit leichter oder mässiger Nierenfunktionsstörung im Vergleich zu Patienten mit normaler Nierenfunktion untersucht. Es wurden keine klinisch bedeutsamen Unterschiede in der Clearance von Nivolumab oder Relatlimab zwischen Patienten mit leichter oder mässiger Einschränkung der Nierenfunktion und Patienten mit normaler Nierenfunktion festgestellt. Für Patienten mit stark eingeschränkter Nierenfunktion liegen nicht genügend Daten vor, um Schlussfolgerungen für diese Patientenpopulation ziehen zu können.
Präklinische Daten
Genotoxizität / Kanzerogenität
Weder mit Nivolumab noch Relatlimab wurden Mutagenitäts- oder Karzinogenitätsstudien durchgeführt. Da es sich um Antikörper handelt, wird keine direkte Interaktion mit der DNA erwartet.
Reproduktionstoxitität
Es wurden keine speziellen Studien zur Reproduktionstoxizität mit Nivolumab in Kombination mit Relatlimab durchgeführt.
Relatlimab
Es liegen keine tierexperimentellen Daten zur Wirkung von Relatlimab auf Trächtigkeit und Fortpflanzung vor. Allerdings wurden die Auswirkungen von murinen Anti-LAG 3-Antikörpern in Mäusen unter Verwendung von syngenen und allogenen Zuchtmodellen untersucht. Anti-LAG-3-Antikörper waren gut verträglich, wenn sie ab dem 6. Trächtigkeitstag verabreicht wurden. Weder bei syngenen noch bei allogenen Züchtungen traten mütterliche oder entwicklungsbezogene Effekte auf. Die Auswirkungen von Relatlimab auf die pränatale und postnatale Entwicklung wurden nicht untersucht. Jedoch kann die Blockade von LAG-3 mit Relatlimab, basierend auf dem Wirkmechanismus, einen ähnlichen negativen Effekt auf die Schwangerschaft haben wie Nivolumab. Es wurden keine Fertilitätsstudien mit Relatlimab durchgeführt.
Nivolumab
Die Blockade des PD-1/PD-L1-Signalwegs hat in Mausmodellen zur Schwangerschaft gezeigt, dass sie die Toleranz gegenüber dem Fetus stört und die Häufigkeit von Fehlgeburten erhöht. Die Auswirkungen von Nivolumab auf die pränatale und postnatale Entwicklung wurden bei Affen untersucht, die vom Beginn der Organogenese im ersten Trimester bis zur Geburt, zweimal wöchentlich Nivolumab erhielten. Dabei waren die Expositionslevel entweder 8- oder 35-mal höher als jene, die bei der klinischen Dosis von 3 mg/kg Nivolumab (basierend auf der AUC) beobachtet wurden. Es gab einen dosisabhängigen Anstieg der Häufigkeit von Fehlgeburten und eine erhöhte neonatale Sterblichkeit ab dem dritten Trimenon.
Die verbleibenden Nachkommen der mit Nivolumab behandelten Weibchen überlebten bis zum planmässigen Abbruch, ohne behandlungsbedingte klinische Symptome, Veränderungen der normalen Entwicklung, Auswirkungen auf das Organgewicht oder makro- und mikroskopische pathologische Veränderungen. Die Ergebnisse für Wachstumsindizes sowie teratogene, neurologische, immunologische und klinisch-pathologische Parameter während des gesamten 6-monatigen postnatalen Zeitraums waren mit denen der Kontrollgruppe vergleichbar. Aufgrund des Wirkmechanismus kann jedoch die fetale Exposition gegenüber Nivolumab und in ähnlicher Weise auch Relatlimab, das Risiko für die Entwicklung immunbezogener Störungen oder die Veränderung der normalen Immunantwort erhöhen. Bei PD-1 und PD-1/LAG-3 Knockout-Mäusen wurde über immunvermittelte Störungen berichtet. Fertilitätsstudien wurden mit Nivolumab nicht durchgeführt.
Sonstige Hinweise
Inkompatibilitäten
Da keine Kompatibilitätsstudien vorliegen, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden. Opdualag sollte nicht gleichzeitig mit anderen Arzneimitteln zusammen über denselben intravenösen Zugang verabreicht werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach dem Öffnen: Aus mikrobiologischer Perspektive muss das Arzneimittel nach dem Öffnen sofort infundiert oder verdünnt und infundiert werden.
Nach der Vorbereitung der Infusion: Die Verabreichung der Opdualag-Infusion muss innerhalb 24 h nach der Vorbereitung abgeschlossen werden. Wenn die Infusionslösung nicht sofort verwendet wird, kann sie im Kühlschrank (2-8°C) und vor Licht geschützt für maximal 24 h gelagert werden (maximal 8 h der gesamten 24 h Lagerung können bei Raumtemperatur (15-25°C) und Raumlicht erfolgen - der maximale Zeitraum von 8 h bei Raumtemperatur und Raumlicht muss den Zeitraum für die Verabreichung des Produkts umfassen).
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Die ungeöffnete Durchstechflasche kann bei kontrollierter Raumtemperatur von bis zu 25°C und bei Raumlicht bis zu 72 h gelagert werden.
Hinweise für die Handhabung
Opdualag wird als Einzeldosis-Durchstechflasche geliefert und enthält keine Konservierungsmittel. Aseptische Techniken müssen eingehalten werden.
Opdualag kann zur intravenösen Verabreichung verwendet werden, entweder:
unverdünnt nach Umfüllen in einen Infusionsbehälter mit einer geeigneten sterilen Spritze oder
nach Verdünnung gemäss folgenden Anweisungen:
-Die endgültige Konzentration der Infusion sollte zwischen 3 mg/ml Nivolumab und 1 mg/ml Relatlimab bis 12 mg/ml Nivolumab und 4 mg/ml Relatlimab liegen.
-Das Gesamtvolumen der Infusion darf 160 ml nicht überschreiten. Bei erwachsenen Patienten unter 40 kg darf das Gesamtvolumen der Infusion 4 ml pro Kilogramm des Patientengewichts nicht überschreiten.
Das Opdualag-Konzentrat kann verdünnt werden mit entweder:
-Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke oder
-Glukoselösung 50 mg/ml (5%) für Injektionszwecke.
Zubereitung der Infusion
-Untersuchen Sie das Opdualag-Konzentrat auf Schwebestoffteilchen oder Verfärbung. Die Durchstechflasche nicht schütteln. Opdualag ist eine klare bis opaleszierende, farblose bis blass gelbe Flüssigkeit. Die Durchstechflasche ist zu entsorgen, wenn die Lösung eine Trübung, Verfärbung oder Schwebstoffteilchen, ausgenommen wenige durchscheinend bis weissliche Partikel, aufweist.
-Ziehen Sie die erforderliche Menge Opdualag-Konzentrat mithilfe einer geeigneten sterilen Spritze auf und füllen Sie das Konzentrat in einen sterilen Infusionsbehälter (Ethylvinylacetat (EVA), Polyvinylchlorid (PVC), oder Polyolefin).
-Verdünnen Sie das Konzentrat gegebenenfalls mit der benötigten Menge Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke oder Glukoselösung 50 mg/ml (5%) für Injektionszwecke. Zur Erleichterung der Zubereitung kann das Konzentrat auch direkt in einen vorgefüllten Beutel überführt werden, der das entsprechende Volumen an Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke oder Glukoselösung 50 mg/ml (5%) für Injektionszwecke enthält.
-Infusionslösung vorsichtig durch manuelle Drehung mischen. Nicht schütteln.
Verabreichung
Die Opdualag-Infusionslösung darf nicht als intravenöse Druck- oder Bolusinjektion verabreicht werden.
Verabreichen Sie die Opdualag-Infusionslösung intravenös über einen Zeitraum von 30 Minuten.
Verwenden Sie ein Infusionsset und einen sterilen, nicht-pyrogenen In-Line- oder Zusatz-Filter mit geringer Proteinbindung (Porengrösse 0,2 μm bis 1,2 μm).
Die Opdualag-Infusionslösung ist mit EVA-, PVC- und Polyolefin-Behältern, PVC-Infusionssets und Inline-Filtern mit Polyethersulfon- (PES), Nylon- und Polyvinylidenfluorid- (PVDF) Membranen mit Porengrössen von 0,2 µm bis 1,2 µm kompatibel.
Verabreichen Sie nicht gleichzeitig andere Arzneimittel über den gleichen intravenösen Zugang.
Spülen Sie nach Verabreichung der Opdualag-Dosis die Leitung mit Natriumchlorid 9 mg/ml (0,9%) Lösung zur Injektion oder 50 mg/ml (5%) Glukoselösung zur Injektion.
Entsorgung
Verbliebene Restmengen der Infusionslösung dürfen nicht zur weiteren Verwendung gelagert werden. Nicht verwendetes Arzneimittel oder Abfallmaterialien sind gemäss den lokalen Bestimmungen zu entsorgen.
Zulassungsnummer
68609 (Swissmedic).
Packungen
Durchstechflasche zu 240 mg/80 mg pro 20 ml: 1 [A]
Zulassungsinhaberin
Bristol-Myers Squibb SA, Steinhausen.
Stand der Information
März 2026