Zusammensetzung
Wirkstoffe
Seladelpar als Seladelpar-Lysin-Dihydrat
Hilfsstoffe
Kapselinhalt: Mikrokristalline Cellulose, Mannitol, Croscarmellose-Natrium, Butylhydroxytoluol, Magnesiumstearat, hochdisperses Siliciumdioxid.
Kapselhülle: Gelatine, Titandioxid, Eisenoxid schwarz (E172), Eisenoxid rot (E172), Eisenoxid gelb (E172), Indigocarmin (E132).
Schwarze Farbe für den Aufdruck "10" (auf dem Kapselunterteil) enthält: Schellack (E904), Propylenglycol (E1520), Kaliumhydroxid (E525), Eisenoxid schwarz (E172).
Weisse Farbe für den Aufdruck "CBAY" (auf dem Kapseloberteil) enthält: Schellack (E904), Propylenglycol (E1520), Natriumhydroxid (E524), Povidon (E1201), Titandioxid.
Eine Hartkapsel Lyvdelzi enthält 0,42 mg Natrium.
Darreichungsform und Wirkstoffmenge pro Einheit
Eine Hartkapsel mit Seladelpar-Lysin-Dihydrat entsprechend 10 mg Seladelpar.
Opake Hartkapseln aus Gelatine mit dunkelblau-opakem Kapseloberteil und hellgrau-opakem Kapselunterteil mit dem Aufdruck "CBAY" in weisser Farbe auf dem Kapseloberteil und "10" in schwarzer Farbe auf dem Kapselunterteil.
Indikationen/Anwendungsmöglichkeiten
Lyvdelzi wird angewendet für die Behandlung der primär biliären Cholangitis (PBC), in Kombination mit Ursodeoxycholsäure (UDCA) bei Erwachsenen, die nicht ausreichend auf UDCA alleine ansprechen, oder als Monotherapie bei Erwachsenen, die UDCA nicht vertragen (siehe "Klinische Wirksamkeit" ).
Diese Indikation wurde auf der Grundlage einer Verringerung der alkalischen Phosphatase (ALP) genehmigt (siehe "Eigenschaften/Wirkungen" ). Eine Verbesserung der Überlebensrate oder die Verhinderung von Leberdekompensationen wurde nicht nachgewiesen.
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen Dokumentation, wird diese Indikation befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine Zulassung ohne besondere Auflagen überführt werden.
Dosierung/Anwendung
Empfohlene Dosierung
Die empfohlene Dosis von Lyvdelzi beträgt 10 mg einmal täglich.
Art der Anwendung
Zum Einnehmen.
Die Hartkapseln können mit einer Mahlzeit oder unabhängig davon eingenommen werden.
Verpasste Dosis
Wenn eine Dosis von Lyvdelzi vergessen wurde, sollte der Patient die nachfolgende Dosis zum nächsten vorgesehenen Zeitpunkt einnehmen. Es sollte keine doppelte Dosis eingenommen werden, um die vergessene Dosis nachzuholen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) ist keine Dosisanpassung von Lyvdelzi erforderlich.
Die Sicherheit und Wirksamkeit von Lyvdelzi bei Patienten mit dekompensierter Zirrhose (Child-Pugh-Klasse B oder C) wurden nicht nachgewiesen. Die Anwendung von Lyvdelzi bei Patienten mit bestehender oder sich entwickelnder dekompensierter Zirrhose wird nicht empfohlen (siehe "Pharmakokinetik" ).
Patienten mit Zirrhose sind auf Anzeichen einer Dekompensation zu überwachen. Bei Patienten mit Fortschreiten zu einer mittelschweren oder schweren Leberfunktionsstörung (Child-Pugh-Klasse B oder C) ist die Behandlung mit Lyvdelzi abzubrechen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter (geschätzte glomeruläre Filtrationsrate [estimated glomerular filtration rate, eGFR] ≥60 bis < 90 ml/min), mittelschwerer (eGFR ≥30 bis < 60 ml/min) oder schwerer (eGFR < 30 ml/min) Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Es liegen keine Studien zu Lyvdelzi bei Patienten mit dialysepflichtiger terminaler Niereninsuffizienz (end stage renal disease, ESRD) vor, so dass für diese Patienten keine Dosierungsempfehlung gegeben werden kann (siehe "Pharmakokinetik" ).
Ältere Patienten
Bei Patienten ab einem Alter von 65 Jahren ist keine Dosisanpassung erforderlich (siehe "Pharmakokinetik" ).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Lyvdelzi bei Kindern und Jugendlichen wurde nicht nachgewiesen.
Lyvdelzi ist für die Anwendung bei Kindern und Jugendlichen nicht zugelassen.
Anpassung der Verabreichung bei Therapie mit Gallensäurebindenden Harzen
Lyvdelzi soll mindestens 4 Stunden vor oder 4 Stunden nach der Einnahme von Gallensäurebindenden Harzen oder in einem möglichst grossen zeitlichen Abstand eingenommen werden (siehe "Interaktionen" ).
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und Vorsichtsmassnahmen
Auffällige Leberwerte
Lyvdelzi wird mit dosisabhängigen Erhöhungen der Serumtransaminasen (Aspartat-Aminotransferase [AST] und Alanin-Aminotransferase [ALT]) um mehr als das 3-Fache der Obergrenze des Normalwerts (upper limit of normal, ULN) bei PBC-Patienten in Verbindung gebracht, die einmal täglich 50 mg (das 5-Fache der empfohlenen Dosis) und einmal täglich 200 mg (das 20-Fache der empfohlenen Dosis) erhielten. Nach dem Absetzen von Lyvdelzi kehrten die Transaminasewerte auf die Werte vor der Behandlung zurück. Für Lyvdelzi 10 mg einmal täglich zeigte sich kein ähnliches Muster für den Anstieg der Transaminasewerte (siehe "Überdosierung" ).
Zu Beginn der Behandlung mit Lyvdelzi sollten klinische und laborchemische Untersuchungen erfolgen und die Werte anschliessend gemäss der routinemässigen klinischen Praxis überwacht werden. Die Behandlung mit Lyvdelzi ist zu unterbrechen, wenn sich die Leberwerte (ALT, AST, Gesamtbilirubin und/oder alkalische Phosphatase [ALP]) verschlechtern oder wenn der Patient Anzeichen und Symptome einer klinischen Hepatitis entwickelt (z.B. Ikterus, Schmerzen im oberen rechten Quadranten, Eosinophilie). Bei einer Verschlechterung der Leberwerte bei Wiederaufnahme der Behandlung mit Lyvdelzi ist ein endgültiger Abbruch der Behandlung in Erwägung zu ziehen.
Biliäre Obstruktion
Bei Patienten mit vollständiger biliärer Obstruktion ist die Anwendung von Lyvdelzi zu vermeiden. Bei Verdacht auf eine biliäre Obstruktion ist die Behandlung mit Lyvdelzi zu unterbrechen und eine Behandlung gemäss klinischer Indikation einzuleiten.
Frakturen
Bei 3,9 % (n=5) der mit Lyvdelzi in der pivotalen Studie CB8025-32048 behandelten Patienten traten Frakturen auf, im Vergleich zu 0 % der mit Placebo behandelten Patienten (siehe "Unerwünschte Wirkungen" ). Berücksichtigen Sie das Frakturrisiko bei Patienten, die mit Lyvdelzi behandelt werden, und überwachen Sie die Knochendichte gemäss den aktuellen Behandlungsstandards
Gleichzeitige Anwendung mit anderen Arzneimitteln
Die gleichzeitige Anwendung von Probenecid zusammen mit Lyvdelzi wird nicht empfohlen (siehe "Interaktionen" ).
Die gleichzeitige Anwendung von Lyvdelzi mit Ciclosporin und Fluconazol zusammen wird nicht empfohlen (siehe "Interaktionen" ).
Hilfsstoffe
Lyvdelzi enthält weniger als 1 mmol Natrium (23 mg) pro Hartkapsel, d.h. es ist nahezu "natriumfrei" .
Interaktionen
Wirkung anderer Arzneimittel auf Seladelpar
Probenecid
Die gleichzeitige Verabreichung von Seladelpar mit Probenecid (einem OAT3- und OATP1B3-Inhibitor) kann die Seladelpar-Exposition erhöhen. Die gleichzeitige Anwendung von Probenecid zusammen mit Seladelpar wird nicht empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
In einer klinischen Studie speziell zu Arzneimittelwechselwirkungen wurde nach der gleichzeitigen Anwendung einer Einzeldosis von 10 mg Seladelpar zusammen mit 500 mg Probenecid bei gesunden Probanden eine Erhöhung der AUC0-inf um das 2-Fache und der Cmax um das 4,69-Fache beobachtet.
Kombination von Arzneistofftransporter und CYP2C9- und CYP3A4-Inhibitoren
Bei gleichzeitiger Anwendung von Seladelpar mit Ciclosporin (einem BCRP-, OATP1B1-, OATP1B3- und CYP3A4-Inhibitor) und Fluconazol (einem moderaten CYP2C9- und CYP3A4-Inhibitor) wird eine starke Erhöhung der Seladelpar Exposition (auf mehr als das 5-Fache) erwartet. Die gleichzeitige Anwendung einer solchen Kombination wird nicht empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Arzneistofftransporter-Inhibitoren
Die gleichzeitige Anwendung von Seladelpar zusammen mit dualen oder multiplen klinischen Inhibitoren von Arzneistofftransportern, einschließlich BCRP, OATP1B1, OATP1B3 und OAT3 (z.B. Ciclosporin), kann zu einem Anstieg der Seladelpar-Exposition führen. Bei gleichzeitiger Anwendung von Seladelpar zusammen mit dualen oder multiplen klinischen Arzneistofftransportern, einschließlich BCRP, OATP1B1, OATP1B3 und OAT3, sollten Patienten engmaschig auf Nebenwirkungen überwacht werden.
In einer klinischen Studie speziell zu Arzneimittelwechselwirkungen wurde nach der gleichzeitigen Anwendung einer Einzeldosis von 10 mg Seladelpar zusammen mit 600 mg Ciclosporin (einem BCRP-, OATP1B1-, OATP1B3- und CYP3A4-Inhibitor) bei gesunden Probanden ein Anstieg der AUC0-inf von Seladelpar um das 2,1-Fache und der Cmax von Seladelpar um das 2,9-Fache beobachtet.
CYP2C9- und CYP3A4-Inhibitoren
Seladelpar wird in vitro vorwiegend über CYP2C9 metabolisiert und in geringerem Masse über CYP2C8 und CYP3A4. Die gleichzeitige Anwendung von Seladelpar zusammen mit Arzneimitteln, die starke CYP2C9-Inhibitoren sind, oder mit dualen moderaten CYP2C9- und moderaten bis starken CYP3A4-Inhibitoren kann zu einem Anstieg der Seladelpar-Exposition führen. Bei gleichzeitiger Anwendung von Seladelpar zusammen mit Arzneimitteln, die starke CYP2C9-Inhibitoren sind (z.B. Fluconazol, Mifepriston) oder mit dualen moderaten CYP2C9- und moderaten bis starken CYP3A4-Inhibitoren sollten Patienten auf Nebenwirkungen überwacht werden.
In einer klinischen Studie speziell zu Arzneimittelwechselwirkungen wurde nach der gleichzeitigen Anwendung einer Einzeldosis von 10 mg Seladelpar zusammen mit 400 mg Fluconazol (einem moderaten CYP2C9- und CYP3A4-Inhibitor) bei gesunden Probanden ein Anstieg der AUC0-inf von Seladelpar um das 2,4-Fache und der Cmax von Seladelpar um das 1,4-Fache beobachtet.
CYP2C9-Induktoren und starke CYP3A4-Induktoren
Die gleichzeitige Anwendung von Seladelpar zusammen mit Arzneimitteln, die CYP2C9-Induktoren und starke CYP3A4-Induktoren sind (z.B. Rifampicin, ein starker CYP3A4- und mässiger CYP2C9-Induktor), kann die Seladelpar-Exposition verringern. Bei gleichzeitiger Anwendung von Seladelpar zusammen mit Arzneimitteln, die CYP2C9-Induktoren und starke CYP3A4-Induktoren sind, sollten Patienten auf eine mögliche Verringerung der Wirksamkeit überwacht werden.
Nach Anwendung von Carbamazepin 300 mg zweimal täglich, gefolgt von einer Einzeldosis von 10 mg Seladelpar an gesunde Probanden nahmen die AUC0-inf von Seladelpar um etwa 44% und die Cmax von Seladelpar um 24% ab. Die Dosis von Carbamazepin (eines starken CYP3A- und schwachen CYP2C9-Induktors) wurde über 7 Tage von 100 mg auf 300 mg gesteigert.
Gallensäurebindende Harze
Gallensäurebindende Harze wie Cholestyramin, Colestipol oder Colesevelam können die Absorption anderer, gleichzeitig angewendeter Arzneimittel verringern. Seladelpar sollte mindestens 4 Stunden vor oder 4 Stunden nach der Einnahme eines gallensäurebindenden Harzes eingenommen werden.
Chinidin
Bei gleichzeitiger Verabreichung einer Einzeldosis von 600 mg Chinidin (einem P-gp-Inhibitor) an gesunde Probanden waren keine signifikanten Veränderungen der Seladelpar-Expositionen zu beobachten.
Wirkung von Seladelpar auf andere Arzneimittel
Seladelpar hat keine klinisch relevante Wirkung auf die Pharmakokinetik von Tolbutamid (CYP2C9-Substrat), Midazolam (CYP3A4-Substrat), Simvastatin (CYP3A4- und OATP-Substrat), Atorvastatin (CYP3A4- und OATP-Substrat) und Rosuvastatin (OATP- und BCRP-Substrat).
In-vitro-Studien
Basierend auf In-vitro-Studien wird erwartet, dass eine Dosis von 10 mg Seladelpar keinen signifikanten Einfluss hat auf die Pharmakokinetik von gleichzeitig verabreichten Arzneimitteln, die Substrate von CYP-Enzymen (1A2, 2B6, 2C8, 2C19, 2D6), UGTs, P-gp, MATEs, OCT1, OCT2, OAT1 oder OAT3 sind.
Seladelpar ist in vitro ein Substrat der Transporter OATP1B1, OATP1B3, BCRP, P-gp und OAT3. Seladelpar ist kein Substrat von MATE1, MATE2-K, OAT1, OCT1 oder OCT2.
Schwangerschaft, Stillzeit
Schwangerschaft
Bisher liegen keine oder nur sehr begrenzte Erfahrungen (weniger als 300 Schwangerschaftsausgänge) mit der Anwendung von Seladelpar bei Schwangeren vor.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf die Reproduktionstoxizität (siehe "Präklinische Daten" ).
Als Vorsichtsmassnahme soll eine Anwendung von Lyvdelzi während der Schwangerschaft vermieden werden.
Stillzeit
Es ist nicht bekannt, ob Seladelpar oder seine Metaboliten in die Muttermilch übergehen.
Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Lyvdelzi verzichtet werden soll / die Behandlung mit Lyvdelzi zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine Daten über die Auswirkung von Seladelpar auf die Fertilität beim Menschen vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte Auswirkungen in Bezug auf die Fertilität oder die Fortpflanzungsfähigkeit (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen von Seladelpar auf die Fahrtüchtigkeit und das Bedienen von Maschinen durchgeführt.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die Beurteilung der Nebenwirkungen basiert auf Daten aus einer pivotalen 52-wöchigen, 2:1 randomisierten, placebokontrollierten klinischen Studie (CB8025-32048) mit 193 PBC-Patienten, von denen 128 Patienten unter Therapie mit 10 mg Seladelpar waren, und supportiv auf einer weiteren vorzeitig beendeten 52-wöchigen Placebo kontrollierten klinischen Studie (CB8025-31735), die sowohl 5 mg als auch 10 mg Seladelpar untersuchte. Insgesamt wurden 217 PBC-Patienten in beiden Studien zusammen mit Seladelpar 10 mg einmal täglich behandelt.
Basierend auf den Erfahrungen aus klinischen Studien waren die am häufigsten berichteten Nebenwirkungen Abdominalschmerz (11,5%), Kopfschmerzen (7,8%), Übelkeit (6,9%), aufgeblähter Bauch (5,1%) und Anämie (3,2%). Diese Nebenwirkungen waren nicht schwerwiegend und führten nicht zum Abbruch der Behandlung mit Seladelpar.
Liste der unerwünschten Wirkungen
Die Nebenwirkungen sind unten in Tabelle 1 nach MedDRA-Systemorganklassen und abnehmender Häufigkeit geordnet. Die Häufigkeiten sind als sehr häufig (≥1/10) und häufig (≥1/100 bis < 1/10) definiert.
Tabelle 1: Tabellarische Auflistung der Nebenwirkungen von Lyvdelzi
Häufigkeita Nebenwirkung Erkrankungen des Blutes und des Lymphsystems Häufig Anämie Erkrankungen des Nervensystems Häufig Kopfschmerzen Erkrankungen des Gastrointestinaltrakts Sehr häufig Abdominalschmerzb (11,5%) Häufig Übelkeit Häufig Aufgeblähter Bauch
a Häufigkeit basierend auf allen Patienten, die Seladelpar 10 mg in CB8025-32048 und CB8025-31735 erhielten.
b Umfasst Abdominalschmerz, Schmerzen im Oberbauch, Schmerzen im Unterbauch und abdominale Beschwerden.
Zusatzinformationen
Frakturen
In der pivotalen Studie CB8025-32048 traten bei 3,9% (n=5) der mit Lyvdelzi behandelten Patienten Frakturen auf, im Vergleich zu 0% der mit Placebo behandelten Patienten. Die Knochenmineraldichte zu Studienbeginn wurde nicht ermittelt. Die mediane Zeit bis zur Fraktur nach Behandlungsbeginn mit Lyvdelzi betrug 295 Tage (Bereich: 89-349).
Laborwertveränderungen
Serumkreatinin
Bei mit Lyvdelzi behandelten Patienten wurde ein dosisabhängiger Anstieg des Serumkreatinins und ein Abfall der eGFR häufiger als bei Placebo behandelten Patienten beobachtet. In der Studie CB8025-32048 wurde bei der Dosis mit 10 mg ein medianer Anstieg des Serumkreatinins von bis zu 6,6% beobachtet, verglichen mit bis zu 2,2% bei Patienten, die Placebo erhielten. Bei 10% (n=12) der mit Lyvdelzi behandelten Patienten kam es zu einem Rückgang der eGFR um mindestens 25%, verglichen mit 2% (n=1) der mit Placebo behandelten Patienten. Bei keinem der Patienten kam es zu einem Rückgang der eGFR um 50% oder mehr. Die Anstiege des Serumkreatinins waren nicht progressiv, und die Werte gingen bei fortgesetzter Behandlung mit Lyvdelzi auf den Ausgangswert mit der Ausnahme von 2 Patienten in der Studie CB8025-32048 wieder zurück. Lyvdelzi musste bei keinem Patienten abgesetzt werden, und es gab keine klinischen Befunde, die mit den beobachteten Veränderungen des Serumkreatinins in Zusammenhang standen.
Meldung des Verdachts auf unerwünschte Wirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Bei PBC-Patienten, die das 5-Fache der empfohlenen Dosis bzw. das 20-Fache der empfohlenen Dosis von Lyvdelzi erhielten, wurden erhöhte Lebertransaminasen, Muskelschmerzen und/oder Erhöhungen der Kreatinphosphokinase beobachtet, die nach dem Absetzen von Lyvdelzi wieder abklangen (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Es gibt keine spezifische Behandlung bei einer Überdosierung mit Lyvdelzi. Gegebenenfalls sind allgemeine unterstützende Massnahmen indiziert. Falls angezeigt, sollte eine Elimination des noch nicht resorbierten Arzneimittels durch Erbrechen oder eine Magenspülung erreicht werden. Dabei sollten die üblichen Vorsichtsmassnahmen zum Freihalten der Atemwege beachtet werden. Da Seladelpar in hohem Masse an Plasmaproteine gebunden wird, ist eine Hämodialyse nicht in Betracht zu ziehen.
Eigenschaften/Wirkungen
ATC-Code
A05AX07
Wirkungsmechanismus
Seladelpar ist ein Agonist des Peroxisom-Proliferator-aktivierten Rezeptors delta (PPARδ) oder Delpar. PPARδ ist ein Kernrezeptor, der in der Leber und in anderen Geweben exprimiert wird. Die Aktivierung von PPARδ verringert die Gallensäuresynthese in der Leber durch die Fibroblasten-Wachstumsfaktor 21 (Fibroblast Growth Factor 21, FGF21)-abhängige Herunterregulierung von CYP7A1, dem wichtigsten Enzym für die Gallensäuresynthese aus Cholesterin, und durch die Verringerung der Cholesterinsynthese und -resorption.
Pruritus ist ein häufiges Symptom bei Patienten mit PBC, die Ursachen sind aber nicht vollständig geklärt. Die Behandlung mit Seladelpar hat eine Reduzierung des Pruritus gezeigt.
Pharmakodynamik
In kontrollierten klinischen Studien führte die Behandlung mit Seladelpar zu einer Reduktion von ALP, einem Biomarker der Cholestase. Eine ALP-Reduktion wurde innerhalb von 1 Woche nach Behandlungsbeginn beobachtet, nahm bis Monat 3 weiter ab und hielt bis Monat 12 an.
Kardiale Elektrophysiologie
Bei dem 20-Fachen der empfohlenen Dosis von 10 mg führte Seladelpar zu keinen klinisch signifikanten Verlängerungen des QTc-Intervalls.
Klinische Wirksamkeit
Studie CB8025-32048 – RESPONSE
Die Wirksamkeit von Lyvdelzi wurde in der 52-wöchigen, randomisierten, doppelblinden, placebokontrollierten Studie CB8025-32048 untersucht. An der Studie nahmen 193 erwachsene Patienten mit PBC teil, mit unzureichendem Ansprechen oder Unverträglichkeit gegenüber UDCA. Patienten wurden in die Studie aufgenommen, wenn ihr ALP-Spiegel ≥1,67 × ULN und ihr Gesamtbilirubin (TB)-Wert ≤2 × ULN betrug. Patienten mit anderen chronischen Lebererkrankungen, klinisch bedeutender hepatischer Dekompensation, einschliesslich portaler Hypertonie mit Komplikationen, oder Zirrhose mit Komplikationen (z.B. Modell für Lebererkrankungen im Endstadium [MELD]-Score 12 oder höher, bekannte Ösophagusvarizen oder Varizenblutungen in der Vorgeschichte, hepatorenales Syndrom in der Vorgeschichte) wurden von der Studie ausgeschlossen.
Die Patienten wurden randomisiert (2:1) und erhielten 12 Monate lang einmal täglich entweder Lyvdelzi (n=128) 10 mg oder Placebo (n=65). In der Studie erhielten 181 (94%) Patienten Lyvdelzi oder Placebo in Kombination mit UDCA, und 12 (6%) Patienten mit einer Unverträglichkeit gegenüber UDCA erhielten Lyvdelzi oder Placebo als Monotherapie.
Die beiden Behandlungsgruppen waren hinsichtlich der demographischen Merkmale und der Krankheitsmerkmale zu Studienbeginn insgesamt ausgewogen. Das mittlere Alter der Patienten betrug 57 Jahre (Spanne: 28 bis 75), 95% waren weiblich, 88% waren Weisse, 6% waren asiatischer Abstammung, 2% waren Schwarze oder Afroamerikaner und 3% waren amerikanische Indianer oder Ureinwohner Alaskas. Insgesamt 29% der Patienten, d.h. 23% im Arm mit Lyvdelzi 10 mg und 42% im Placebo-Arm, waren hispanischer/lateinamerikanischer Abstammung.
Bei Studienbeginn erfüllten 18 (14%) der mit Lyvdelzi und 9 (14%) der mit Placebo behandelten Patienten mindestens eines der folgenden Kriterien für eine Zirrhose: Fibroscan > 16,9 kPa, Biopsie oder radiologischer Befund in der Vorgeschichte mit Hinweis auf eine Zirrhose, Thrombozytenzahl < 140'000/μl mit mindestens einem zusätzlichen Laborbefund, darunter Serumalbumin < 3,5 g/dl, INR > 1,3 oder Gesamtbilirubin > 1 × ULN oder klinische Feststellung einer Zirrhose durch den Prüfarzt bzw. die Prüfärztin. Alle Zirrhose-Patienten hatten bei Studienbeginn einen Child-Pugh-Klasse A Status.
Die mittlere ALP-Konzentration zu Studienbeginn betrug 314 Einheiten pro Liter (E/l) (Spanne: 161 bis 786), entsprechend 2,7 × ULN. Die mittlere Konzentration an Gesamtbilirubin zu Studienbeginn betrug 0,8 mg/dl (Spanne: 0,3 bis 1,9) und war bei 87% der Patienten ≤ ULN. Weitere mittlere biochemische Leberwerte zu Studienbeginn betrugen: 48 E/l (Spanne: 9 bis 115) für ALT, entsprechend dem 1,2 × ULN, 40 E/l (Spanne: 16 bis 94) für AST, entsprechend dem 1,2 × ULN, und 288 E/l (Spanne: 42 bis 1088) für Gamma-Glutamyltransferase (GGT), entsprechend dem 1,7 × ULN.
Lyvdelzi zeigte eine signifikant grössere Verbesserung des biochemischen Ansprechens und der ALP-Normalisierung in Monat 12 im Vergleich zu Placebo. Die Behandlung mit Lyvdelzi führte zu einem signifikant höheren Prozentsatz von Patienten (62%), die den primären Wirksamkeitsendpunkt eines kombinierten biochemischen Ansprechens in Monat 12 im Vergleich zu Placebo (20%) erreichten (p < 0,0001). Der ULN für ALP wurde mit 116 U/L definiert. Der ULN für Gesamtbilirubin wurde mit 1,1 mg/dL definiert. Tabelle 2 zeigt die Ergebnisse nach 12 Monaten für den Prozentsatz der Patienten, die ein biochemisches Ansprechen, jede Komponente des biochemischen Ansprechens und eine ALP-Normalisierung erreichten. Insgesamt hatten 87% der Patienten bei Studienbeginn eine TB-Konzentration von weniger als oder gleich ULN. Daher war die Verbesserung des ALP der Hauptfaktor für die Ergebnisse der biochemischen Ansprechrate nach 12 Monaten.
Tabelle 2: Studie CB8025-32048: Kombinierter biochemischer Endpunkt und ALP-Normalisierung mit Lyvdelzi mit oder ohne UDCA
Lyvdelzi 10 mg Placebo(n=65) Differenz zwischen den
(n=128) Behandlungen % (95%-KI)d
Primärer kombinierter
Endpunkt in Monat 12
Biochemische Ansprechrate, 79 (62) [53; 70] 13 (20) [10; 30] 42 (28; 53) p < 0,0001
n (%)a, b [95%-KI]
Komponenten des biochemisch
en Ansprechens
ALP < 1,67 × ULN, n (%) 84 (66) 17 (26) 39 (25; 52)
ALP-Verringerung um 107 (84) 21 (32) 51 (37; 63)
mindestens 15%, n (%)
Gesamtbilirubin ≤ ULN, n 104 (81) 50 (77) 4 (-7; 17)
(%)
ALP-Normalisierung, n 32 (25) [18; 33] 0 (0) [0; 0] 25 (18; 33) p < 0,0001
(%)b, c [95%-KI]
Patienten, welche die Behandlung vor Monat 12 abbrachen oder Patienten mit fehlenden Daten wurden als Non-Responder eingestuft.
a Biochemisches Ansprechen definiert als ALP-Wert < 1,67 × ULN, ALP-Abnahme ≥15% und Gesamtbilirubin ≤ ULN.
b Berechnung der p-Werte anhand des Cochran-Mantel-Haenszel-Tests, stratifiziert nach ALP-Spiegel zu Studienbeginn (< 350 E/l versus ≥350 E/l) und Pruritus-Score auf der numerischen Rating-Skala (NRS) zu Studienbeginn (< 4 versus ≥4).
c ALP-Normalisierung definiert als ALP ≤ ULN.
d Die 95%-Konfidenzintervalle (KI) wurden basierend auf der Methode von Miettinen und Nurminen ohne Stratifizierung angegeben.
Biochemische Leberwerte
Abbildung 1 zeigt die mittleren ALP-Verringerungen bei mit Lyvdelzi behandelten Patienten im Vergleich zu Placebo über 12 Monate. Verringerungen wurden in Monat 1 beobachtet, setzten sich bis Monat 6 fort und hielten bis Monat 12 an. Die mittlere Veränderung (SE) der kleinsten Quadrate (LS) gegenüber dem Ausgangswert der ALP in Monat 12 betrug in der Gruppe mit Lyvdelzi 10 mg -134 (-151, -117) E/l und in der Placebo-Gruppe -17 (-40, 6) E/l.
Abbildung 1. ALP-Veränderung ab Studienbeginn über 12 Monate in der Studie CB8025-32048 nach Behandlungsarm mit oder ohne UDCAa
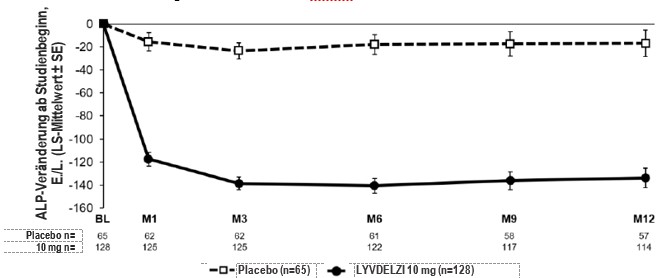
a In der Studie CB8025-32048 hatten 12 Patienten (6%) eine Unverträglichkeit gegenüber UDCA und erhielten die Behandlung als Monotherapie: 8 Patienten (6%) im Arm mit LYVDELZI 10 mg und 4 Patienten (6%) im Placebo-Arm.
In der Subgruppe von Patienten mit ALP < 350 E/l zu Studienbeginn erreichten 76% (71/93) bzw. 23% (11/47) Patienten im Arm mit Lyvdelzi 10 mg bzw. im Placebo-Arm in Monat 12 ein Ansprechen. Von den Patienten mit ALP ≥350 E/l zu Studienbeginn erreichten 23% (8/35) bzw. 11% (2/18) im Arm mit Lyvdelzi 10 mg bzw. im Placebo-Arm in Monat 12 ein Ansprechen.
Pruritus
Bei Patienten mit durchschnittlichen Pruritus-Scores von ≥4 zu Studienbeginn, gemessen anhand des Pruritus-NRS-Scores, führte Lyvdelzi im Vergleich zu Placebo in Monat 6 zu einer signifikanten Verringerung des Pruritus, einem sekundären Schlüssel-Endpunkt in der Studie CB8025-32048 (siehe Tabelle 3). Ein einzelner, von Patienten gemeldeter Endpunkt, die Numerische Rating-Skala (NRS) für Pruritus, bewertete in Studie CB8025-32048 die täglich schlimmste Juckreizintensität der Patienten auf einer 11-stufigen Bewertungsskala mit Werten von 0 ( "kein Juckreiz" ) bis 10 ( "schlimmster vorstellbarer Juckreiz" ). Die NRS für Pruritus wurde täglich in einer 14-tägigen Anlaufphase vor der Randomisierung bis zum 6. Monat durchgeführt.
Tabelle 3 zeigt die Ergebnisse des Vergleichs zwischen Lyvdelzi und Placebo bei diesem Endpunkt, der die Veränderung des Pruritus-Scores gegenüber dem Ausgangswert nach 6 Monaten bei Patienten mit einem durchschnittlichen Pruritus-Score von mindestens 4 zu Studienbeginn bewertet. Der durchschnittliche Pruritus-Score zu Studienbeginn für jeden Patienten wurde durch Mittelwertbildung der Pruritus-NRS-Scores berechnet, die in der Anlaufphase und an Tag 1 vor Behandlungsbeginn erhoben wurden. Der Pruritus-Score nach 6 Monaten wurden für jeden Patienten durch Mittelwertbildung der Pruritus-NRS-Scores innerhalb der letzten Woche des Monats berechnet. Bei den mit Lyvdelzi behandelten Patienten wurde eine grössere Verbesserung des Pruritus im Vergleich zu Placebo festgestellt.
Tabelle 3: Veränderung des Pruritus-Scores von Studienbeginn bis Monat 6 bei PBC-Patienten in der Studie CB8025-32048 mit einem durchschnittlichen Pruritus-Score von ≥4 zu Studienbeginna
Lyvdelzi 10 mg Placebo(n=23) Differenz zwischen den
(n=49) Behandlungen % (95%-KI)
Durchschnittlicher 6,1 (1,4) 6,6 (1,4) -
Pruritus-Score zu
Studienbeginn, Mittelwert
(SD)b
Veränderung des Pruritus-Sc
ores von Studienbeginn bis
Monat 6c
Mittelwert (SE) -3,2 (0,28) -1,7 (0,41) -1,5 (-2,5; -0,5) p <0,005
a Bewertet anhand der Pruritus-NRS, mit der die Intensität des täglichen Juckreizes der Patienten in seiner schlimmsten Ausprägung auf einer 11-Punkte-Skala von 0 ( "Kein Juckreiz" ) bis 10 ( "Schlimmster vorstellbarer Juckreiz" ) beurteilt wird. Die Pruritus-NRS wurde im Rahmen einer ≥14-tägigen Anlaufphase vor der Randomisierung bis Monat 6 täglich ermittelt. Mittelschwerer bis schwerer Pruritus war definiert als Pruritus-NRS-Score ≥4.
b Der Wert zu Studienbeginn schloss den Mittelwert aller während der Anlaufphase und an Tag 1 täglich berichteten Scores ein. Die Pruritus-Scores wurden für jeden Patienten für die Monate nach Studienbeginn durch Mittelung der Pruritus-NRS-Scores innerhalb der für jeden Monat vorgesehenen Woche berechnet.
c Basierend auf den LS-Mittelwerten aus einem gemischten Modell mit Messwiederholungen (Mixed-Effect Model for Repeated Measures, MMRM) für die Veränderung gegenüber Studienbeginn in den Monaten 1 (Woche 4), 3 (Woche 12) und 6 (Woche 26) unter Berücksichtigung des durchschnittlichen Pruritus-Scores zu Studienbeginn, des ALP-Spiegels zu Studienbeginn (< 350 E/L versus ALP-Spiegel ≥350 E/L), des Behandlungsarms, der Zeit (in Monaten) und der Interaktion zwischen Behandlung und Zeit.
Pharmakokinetik
Absorption
Nach oraler Gabe einer Einzeldosis von 10 mg Lyvdelzi betrug die mediane Zeit bis zum Erreichen der Spitzenkonzentration (Tmax) von Seladelpar 1,5 Stunden. Die systemische Exposition gegenüber Seladelpar stieg von 2 mg (dem 0,2-Fachen der empfohlenen Dosis) auf 15 mg (dem 1,5-Fachen der empfohlenen Dosis) dosisproportional und bei höheren Dosen überproportional an. Bei einer Dosiserhöhung von 10 mg auf 200 mg (das 20-Fache der empfohlenen Dosis) stiegen die mittlere Cmax und die mittlere AUC für Seladelpar um das 70- bzw. 27-Fache an.
Nach einmal täglicher Gabe wurde der Steady-State von Seladelpar ab Tag 4 erreicht, und der Anstieg der AUC betrug weniger als 30%. Bei PBC-Patienten betrug die mittlere (CV) Cmax bzw. AUC für Seladelpar nach einmal täglicher Gabe von 10 mg im Steady-State jeweils 90,5 (42,5%) ng/ml bzw. 817 (44%) ng*h/ml.
Bei gleichzeitiger Anwendung von Seladelpar zusammen mit einer Mahlzeit verzögerte sich die Tmax um 2,5 Stunden im Vergleich zu nüchternen Bedingungen, und die Cmax von Seladelpar reduzierte sich um etwa 32%. Da die Gesamtexposition (AUC) ähnlich ist, wird der Einfluss von Nahrung auf die Pharmakokinetik von Seladelpar nicht als klinisch relevant angesehen.
Distribution
Das scheinbare Verteilungsvolumen von Seladelpar im Steady-State bei PBC-Patienten liegt bei etwa 110,3 l. Die Plasmaproteinbindung von Seladelpar ist grösser als 99 %.
Metabolismus
In vitro wird Seladelpar vorwiegend über CYP2C9 metabolisiert und in geringerem Masse über CYP2C8 und CYP3A4, was zu den drei Hauptmetaboliten führt: Seladelparsulfoxid (M1), Desethyl-Seladelpar (M2) und Desethyl-Seladelparsulfoxid (M3). Das Verhältnis der AUC zwischen Metabolit und Muttersubstanz betrug für M1, M2 bzw. M3 jeweils 0,36, 2,32 und 0,63. Die mediane Tmax für die Metaboliten betrug 10 Stunden für M1 und jeweils 4 Stunden für M2 und M3. Es ist nicht davon auszugehen, dass M1, M2 und M3 klinisch relevante pharmakologische Auswirkungen haben.
Elimination
Die scheinbare orale Clearance von Seladelpar bei PBC-Patienten beträgt 12,6 l/h. Nach Gabe einer Einzeldosis von 10 mg Seladelpar an gesunde Probanden betrug die mittlere Eliminationshalbwertszeit für Seladelpar 6 Stunden. Die Halbwertszeit für Seladelpar lag bei PBC-Patienten im Bereich von 3,8 bis 6,7 Stunden.
Seladelpar wird hauptsächlich als Metaboliten mit dem Urin ausgeschieden. Nach oraler Gabe einer Einzeldosis von 10 mg radioaktiv markiertem Seladelpar beim Menschen wurden innerhalb von 216 Stunden etwa 73,4% (weniger als 0,01% unverändert) der Dosis im Urin und 19,5% (2,02% unverändert) in den Fäzes wiedergefunden. Eine Tierstudie legt ausserdem nahe, dass Seladelpar biliär ausgeschieden wird.
Kinetik spezieller Patientengruppen
Alter, Gewicht, Geschlecht und ethnische Zugehörigkeit
Bezüglich Alter (19 bis 79 Jahre), Gewicht (45,8 bis 127,5 kg), Geschlecht und ethnischer Zugehörigkeit (weiss, schwarz, asiatische Abstammung, sonstige) wurden keine klinisch bedeutsamen Unterschiede in der Pharmakokinetik von Seladelpar festgestellt.
Leberfunktionsstörungen
In einer klinischen pharmakologischen Studie an Teilnehmern mit leichter, mittelschwerer bzw.
schwerer Leberfunktionsstörung verschiedener Ursachen (Child-Pugh-Klasse A, B bzw. C) stieg die AUC von Seladelpar im Vergleich zu Teilnehmern mit normaler Leberfunktion um das 1,10-, 2,52- bzw. 2,12-Fache und die Cmax um das 1,33-, 5,19- bzw. 5,03-Fache an.
In einer weiteren Studie waren die Seladelpar-Expositionen (Cmax, AUC) nach Anwendung einer oralen Einzeldosis von 10 mg Seladelpar bei PBC-Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) mit portaler Hypertonie 1,7- bis 1,8-mal und bei PBC-Patienten mit mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse B) 1,6- bis 1,9-mal höher als bei PBC-Patienten mit leichter Leberfunktionsstörung ohne portale Hypertonie.
Bei PBC-Patienten mit leichter Leberfunktionsstörung und portaler Hypertonie sowie bei PBC-Patienten mit mittelschwerer Leberfunktionsstörung lagen die Akkumulationsraten nach einmal täglicher Gabe von 10 mg Seladelpar über 28 Tage unter dem 1,2-Fachen.
Nierenfunktionsstörungen
In einer speziellen klinischen Studie an Patienten mit leichter (eGFR ≥60 bis < 90 ml/min), mittelschwerer (eGFR ≥30 bis < 60 ml/min) bzw. schwerer (< 30 ml/min, nicht dialysepflichtig) Nierenfunktionsstörung lag die AUC0-inf von Seladelpar nach Gabe einer Einzeldosis von 10 mg Seladelpar um 48%, 33% bzw. 3% über der von Patienten mit normaler Nierenfunktion. Bei Patienten mit Nierenfunktionsstörung war die Cmax von Seladelpar ähnlich der von Patienten mit normaler Nierenfunktion. Diese Unterschiede in der AUC0-inf von Seladelpar werden als nicht klinisch bedeutsam angesehen.
Die Pharmakokinetik von Seladelpar bei hämodialysepflichtigen Patienten wurde nicht untersucht.
Genetische Polymorphismen
Seladelpar wird vorwiegend über das polymorphe Enzym CYP2C9 metabolisiert. Nach einer Einzeldosis von Seladelpar (1 mg bis 25 mg) war die Plasmaexposition von Seladelpar (Dosis-normalisierte AUC0-inf) bei intermediären CYP2C9-Metabolisierern (*1/*2, *1/*8, *1/*3, *2/*2, n=28) um 18% höher als bei normalen CYP2C9-Metabolisierern (*1/*1, n=84). Für langsame Metabolisierer konnte keine Schlussfolgerung gezogen werden, da nur zwei Teilnehmer mit *2/*3 und kein Teilnehmer mit *3/*3 identifiziert wurden. Bei den beiden Teilnehmer mit *2/*3, stieg die Dosis-normalisierte AUC0-inf im Vergleich zu normalen CYP2C9-Metabolisierern um 47% an.
Präklinische Daten
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Verabreichung, Genotoxizität, Kanzerogenität oder zur Reproduktions- und Entwicklungstoxizität, decken präklinische Daten keine besondere Gefahr für den Menschen auf.
Reproduktions- und Entwicklungstoxizität
Seladelpar hatte bei männlichen und weiblichen Ratten bei oralen Dosen von bis zu 100 mg/kg/Tag (eine Exposition, die dem 223-Fachen bzw. 95-Fachen der klinischen AUC bei der empfohlenen täglichen Dosis von 10 mg entspricht) keine Auswirkungen auf die Fertilität oder die Reproduktionsfunktion.
Bei trächtigen Ratten führte Seladelpar in oralen Dosen von bis zu 100 mg/kg/Tag (eine Exposition, die dem 145-Fachen der klinischen AUC entspricht) zu keinen nachteiligen Auswirkungen auf die embryofetale Entwicklung. Bei trächtigen Kaninchen führten orale Dosen von 40 mg/kg/Tag (eine Exposition, die dem 41-Fachen der klinischen AUC entspricht) zu einem verringerten Gewicht der graviden Gebärmutter (möglicherweise aufgrund maternaler Toxizität) und einem verringerten Körpergewicht des Fötus, aber es wurden keine Missbildungen oder embryofetale Letalität beobachtet. Bei 10 mg/kg/Tag (eine Exposition, die dem 2-Fachen der klinischen AUC entspricht) wurden keine nachteiligen Auswirkungen auf die embryofetale Entwicklung festgestellt.
Nach oraler Seladelpar-Gabe an Ratten während der Trächtigkeit und Laktation in Dosen von 0, 5, 20 oder 100 mg/kg/Tag wurde eine dosisabhängige Reduktion des Körpergewichts der Jungtiere während des Zeitraums vor der Entwöhnung beobachtet, die bei 100 mg/kg/Tag mit einer leicht verminderten Überlebensrate vor der Entwöhnung assoziiert war. Es wurden wachstumsbezogene Verzögerungen der Entwicklungsmeilensteine beobachtet (Augenöffnung und Ablösung der Ohrmuschel bei ≥5 mg/kg/Tag, Haarwachstum und sexuelle Reife bei 100 mg/kg/Tag). Wachstumsminderungen setzten sich bei 100 mg/kg/Tag in der Reifephase nach der Entwöhnung fort und wurden als nachteilige Effekte eingestuft. Die Exposition bei 100 mg/kg/Tag betrug das 145-Fache der klinischen AUC. Bei 20 mg/kg/Tag, der Dosierung ohne nachteilige Effekte auf die Nachkommenschaft, entsprach sie dem 15-Fachen der klinischen AUC.
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Für Kinder unzugänglich aufbewahren.
Nicht über 30°C lagern.
Zulassungsnummer
70063 (Swissmedic)
Packungen
Lyvdelzi 10 mg: 30 Hartkapseln [A]
Zulassungsinhaberin
Gilead Sciences Switzerland Sàrl, Zug
Stand der Information
August 2025