Zusammensetzung
Wirkstoffe
Tovorafenib
Hilfsstoffe
Pulver zur Herstellung einer Suspension zum Einnehmen: Erdbeeraroma (enthält Maltodextrin, Triacetin, künstliches Aroma), hochdisperses Siliciumdioxid, Copovidon K-28, Maltodextrin, Mannitol (E421), mikrokristalline Cellulose, Simeticon, Natriumlaurylsulfat, Sucralose
Eine Flasche enthält maximal 2,5 mg Natrium.
Filmtabletten: hochdisperses Siliciumdioxid, Copovidon K-28, Croscarmellose-Natrium, Magnesiumstearat, mikrokristalline Cellulose, orangefarbener Filmüberzug (Hypromellose, Macrogol 8000, Titandioxid (E171), Eisenoxid gelb (E172), Eisenoxid rot (E172)).
Eine Filmtablette enthält maximal 8,46 mg Natrium.
Darreichungsform und Wirkstoffmenge pro Einheit
Pulver zur Herstellung einer Suspension zum Einnehmen: Eine Flasche enthält nach Rekonstitution 300 mg Tovorafenib. 1 ml rekonstituierte Suspension enthält 25 mg Tovorafenib. Weisses bis gebrochen weisses Pulver.
Filmtabletten mit sofortiger Wirkstofffreisetzung: Eine Filmtablette enthält 100 mg Tovorafenib. Filmtabletten, ovale Tabletten mit der Prägung "100" auf der einen Seite und "D101" auf der anderen Seite.
Indikationen/Anwendungsmöglichkeiten
OJEMDA ist indiziert für die Behandlung von Patienten ab 6 Monaten mit pädiatrischem niedriggradigem Gliom (LGG) mit einer BRAF-Fusion oder einem BRAF-Rearrangement oder einer BRAF-V600-Mutation, welches nach einer oder mehreren systemischen Therapien progredient war.
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen Dokumentation wurde diese Indikation befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine Zulassung ohne besondere Auflagen überführt werden.
Dosierung/Anwendung
Die Behandlung mit Tovorafenib muss von einem qualifizierten Arzt bzw. einer qualifizierten Ärztin, der/die Erfahrung mit der Anwendung von Krebsmedikamenten hat, eingeleitet und überwacht werden.
Vor der Einnahme von Tovorafenib muss durch einen validierten Test bestätigt werden, dass bei den Patienten eine BRAF-Fusion, ein BRAF-Rearrangement oder eine BRAF-V600-Mutation vorliegt.
Dosierung
Die empfohlene Dosis von Tovorafenib, bezogen auf die Körperoberfläche (body surface area, BSA), beträgt 380 mg/m2 oral einmal wöchentlich (die empfohlene Höchstdosis beträgt 600 mg oral einmal wöchentlich).
Tovorafenib kann als Tablette mit sofortiger Wirkstofffreisetzung (siehe Tabelle 1) oder als Suspension zum Einnehmen (siehe Tabelle 2) verabreicht werden.
Für Patienten mit einer Körperoberfläche unter 0,3 m2 wurde keine empfohlene Dosis ermittelt.
Tabelle 1: Empfohlene Dosis, bezogen auf die Körperoberfläche (Tabletten)
Körperoberfläche Empfohlene Dosis 0,30–0,89 m2 Die Suspension zum Einnehmen einmal wöchentlich verabreichen (siehe Tabelle 2) 0,90–1,12 m2 400 mg einmal wöchentlich 1,13–1,39 m2 500 mg einmal wöchentlich ≥1,40 m2 600 mg einmal wöchentlich
Tabelle 2: Empfohlene Dosis, bezogen auf die Körperoberfläche (Suspension zum Einnehmen)
Körperoberfläche Volumen pro Dosis* Empfohlene Dosis 0,30–0,35 m2 5 ml 125 mg einmal wöchentlich 0,36–0,42 m2 6 ml 150 mg einmal wöchentlich 0,43–0,48 m2 7 ml 175 mg einmal wöchentlich 0,49–0,54 m2 8 ml 200 mg einmal wöchentlich 0,55–0,63 m2 9 ml 225 mg einmal wöchentlich 0,64–0,77 m2 11 ml 275 mg einmal wöchentlich 0,78–0,83 m2 12 ml 300 mg einmal wöchentlich 0,84–0,89 m2 14 ml 350 mg einmal wöchentlich 0,90–1,05 m2 15 ml 375 mg einmal wöchentlich 1,06–1,25 m2 18 ml 450 mg einmal wöchentlich 1,26–1,39 m2 21 ml 525 mg einmal wöchentlich ≥1,40 m2 24 ml 600 mg einmal wöchentlich
*Die Höchstdosis je Flasche beträgt 300 mg (12 ml).
Behandlungsdauer
Die wöchentliche Verabreichung ist bis zur Krankheitsprogression, zum Verlust des klinischen Nutzens oder zum Auftreten unzumutbarer Toxizität fortzusetzen (siehe Rubrik "Klinische Wirksamkeit" ).
Versäumte oder verspätete Einnahmen
Liegt der versäumte Einnahmezeitpunkt höchstens 3 Tage zurück, ist die versäumte Dosis so schnell wie möglich einzunehmen, und die nächste Dosis ist am nächsten planmässigen Tag einzunehmen.
Sind seit der versäumten Einnahme mehr als 3 Tage verstrichen, ist die versäumte Dosis zu überspringen, und die nächste Dosis ist am nächsten planmässigen Tag einzunehmen.
Der Zeitabstand zwischen den Dosen muss mindestens vier Tage betragen.
Erbrechen
Wenn Erbrechen unmittelbar nach Einnahme der Dosis auftritt, soll die Dosis noch einmal eingenommen werden.
Änderungen der Dosis
Für die Handhabung von unerwünschten Wirkungen kann eine Dosisreduktion, eine Unterbrechung der Behandlung oder ein Absetzen der Behandlung erforderlich sein.
Die empfohlenen Dosisreduktionen aufgrund unerwünschter Wirkungen sind für Tovorafenib Tabletten in Tabelle 3 und für die Tovorafenib Suspension zum Einnehmen in Tabelle 4 angegeben.
Tabelle 3: Empfohlene Dosisreduktionen aufgrund unerwünschter Wirkungen (Tabletten)
Körperoberfläche Erste Dosisreduktion Zweite Dosisreduktion
0,30–1,12 m2 Die Suspension zum Einnehmen einmal
wöchentlich verabreichen (siehe
Tabelle 4)
1,13–1,39 m2 400 mg einmal wöchentlich Die Suspension zum Einnehmen einmal
wöchentlich verabreichen (siehe
Tabelle 4)
≥1,40 m2 500 mg einmal wöchentlich 400 mg einmal wöchentlich
Tabelle 4: Empfohlene Dosisreduktionen aufgrund unerwünschter Wirkungen (Suspension zum Einnehmen)
Körperoberfläche Erste Dosisreduktion Zweite Dosisreduktion
Volumen Dosis Volumen Dosis
0,30–0,35 m2 4 ml 100 mg 3 ml 75 mg
0,36–0,42 m2 5 ml 125 mg 4 ml 100 mg
0,43–0,48 m2 6 ml 150 mg 5 ml 125 mg
0,49–0,54 m2 7 ml 175 mg 6 ml 150 mg
0,55–0,63 m2 8 ml 200 mg 6 ml 150 mg
0,64–0,77 m2 9 ml 225 mg 8 ml 200 mg
0,78–0,83 m2 10 ml 250 mg 8 ml 200 mg
0,84–0,89 m2 12 ml 300 mg 10 ml 250 mg
0,90–1,05 m2 13 ml 325 mg 11 ml 275 mg
1,06–1,25 m2 15 ml 375 mg 13 ml 325 mg
1,26–1,39 m2 18 ml 450 mg 15 ml 375 mg
≥1,40 m2 20 ml 500 mg 16 ml 400 mg
Die empfohlenen Dosisänderungen aufgrund unerwünschter Wirkungen für Tovorafenib sind in Tabelle 5 angegeben.
Tabelle 5: Empfohlene Dosisänderungen aufgrund unerwünschter Wirkungen
Schweregrad der Dosisänderungb
unerwünschten
Arzneimittelwirkunga
Blutung und intratum
orale Blutung
-Unverträglich Grad Behandlung aussetzen. -Falls eine Besserung bis zu Grad 0-1 eintritt, mit
2 -Grad 3 reduzierter Dosis fortsetzen. -Falls keine Besserung eintritt, dauerhaftes
Absetzen der Behandlung erwägen.
-Erstes Auftreten Behandlung aussetzen. -Falls eine Besserung bis zu Grad 0-1 eintritt, mit
eines Grades 4 reduzierter Dosis fortsetzen. -Falls keine Besserung eintritt, dauerhaftes
Absetzen der Behandlung erwägen.
-Rezidivierend Grad Behandlung dauerhaft absetzen.
4
Hauttoxizität,
einschliesslich
Photosensitivität
-Unverträglich Grad Behandlung aussetzen. -Falls eine Besserung bis Grad 0-1 eintritt, mit
2 -Grad 3 oder 4 reduzierter Dosis fortsetzen. -Falls keine Besserung eintritt, dauerhaftes
Absetzen der Behandlung erwägen.
Leberbezogene
Ereignisse
-Grad 3 AST oder Behandlung aussetzen. Falls eine Besserung bis zu Grad ≤2 oder bis zum
ALT -Grad 3 Bilirubi Zustand wie bei Studienbeginn, wie folgt fortsetzen: -Falls die
n Laborwertanomalien innerhalb von 8 Tagen abklingen, die Behandlung mit der
gleichen Dosis fortsetzen. -Falls die Laborwertanomalien nicht innerhalb von
8 Tagen abklingen, die Behandlung mit reduzierter Dosis fortsetzen.
-Erstes Auftreten Behandlung aussetzen. -Falls eine Besserung bis Grad 0-1 eintritt, mit
jeglicher Grad 4 reduzierter Dosis fortsetzen. -Falls keine Verbesserung eintritt, dauerhaftes
Ereignisse Absetzen der Behandlung erwägen.
-Rezidivierend Grad Behandlung dauerhaft absetzen.
4
Andere unerwünschte
Wirkungen
-Unverträglich Grad Behandlung aussetzen. -Falls eine Besserung bis Grad 0-1 eintritt, mit
2 -Grad 3 reduzierter Dosis fortsetzen. -Falls keine Besserung eintritt, dauerhaftes
Absetzen der Behandlung erwägen.
-Grad 4 Behandlung aussetzen. -Falls eine Besserung bis Grad 0-1 eintritt, mit
reduzierter Dosis fortsetzen. -Falls keine Besserung eintritt, dauerhaftes
Absetzen der Behandlung erwägen.
a National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 5.0.
b Die empfohlenen Dosisreduktionen sind in Tabelle 3 und Tabelle 4 zu finden.
Besondere Patientengruppen
Leberfunktionsstörung
Für Patienten mit leichter Leberfunktionsstörung (Bilirubin ≤ obere Normgrenze [upper limit of normal, ULN] und Aspartataminotransferase [AST] > ULN oder Bilirubin > 1x bis 1,5x ULN und beliebiger AST-Wert) wird keine Dosisanpassung empfohlen. Tovorafenib wurde bei Patienten mit mittelschwerer (Bilirubin > 1,5x bis 3x ULN und beliebiger AST-Wert) oder schwerer (Bilirubin > 3x ULN und beliebiger AST-Wert) Leberfunktionsstörung nicht untersucht (siehe Rubrik "Pharmakokinetik" ). Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung sollten während der Behandlung mit Tovorafenib sorgfältig überwacht werden.
Nierenfunktionsstörung
Für Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung (eGFR ≥30 ml/min/1,73 m2, berechnet mit der Schwartz-Formel oder der MDRD-Formel) wird keine Dosisanpassung empfohlen. Tovorafenib wurde bei Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) nicht untersucht (siehe Rubrik "Pharmakokinetik" ).
Pädiatrische Population
Die Sicherheit und Wirksamkeit von Tovorafenib bei Kindern unter 6 Monaten oder mit einer Körperoberfläche < 0.3 m2 wurde nicht in klinischen Studien untersucht. Für Kinder in einem Alter zwischen 6 Monaten und 2 Jahren liegen nur begrenzte Daten zur Sicherheit und Wirksamkeit vor (siehe Rubrik "Unerwünschte Wirkungen" und "Klinische Wirksamkeit" ).
Anwendung
OJEMDA ist zur oralen Anwendung bestimmt. Die Filmtabletten und das Pulver zur Herstellung einer Suspension zum Einnehmen sind austauschbar.
Patienten, die nicht schlucken können oder deren BSA weniger als 0,9 m2 beträgt, sollte die Suspension zum Einnehmen zur Verfügung gestellt werden. Wenn der Patient nicht schlucken kann und eine nasogastrische Sonde in situ hat, kann die OJEMDA Suspension zum Einnehmen über die Sonde verabreicht werden.
OJEMDA kann unabhängig von Mahlzeiten eingenommen werden (siehe Rubrik "Pharmakokinetik" ) und ist regelmässig einmal wöchentlich zur selben Zeit einzunehmen.
OJEMDA ist pädiatrischen Patienten unter der Aufsicht eines Erwachsenen zu verabreichen.
Tabletten:
Die Tabletten müssen im Ganzen mit Wasser geschluckt und dürfen nicht zerkaut, geteilt oder zerkleinert werden.
Pulver zur Herstellung einer Suspension zum Einnehmen:
OJEMDA muss vor der Verabreichung zu der Suspension zum Einnehmen rekonstituiert werden. Vor der erstmaligen Anwendung der Suspension zum Einnehmen sind die Betreuungspersonen (und gegebenenfalls die Patienten) über die ordnungsgemässe Zubereitung, Dosierung und Anwendung von OJEMDA zu instruieren.
Zum Rekonstituieren der Suspension zum Einnehmen wird jeder gelieferten Flasche mit Pulver exakt 14 ml Wasser mit Raumtemperatur zugegeben. Nach der Rekonstitution enthält jede Flasche der Suspension zum Einnehmen 300 mg Tovorafenib in 12 ml (25 mg/ml).
Für Dosen von mehr als 300 mg sind zwei Flaschen zu rekonstituieren, um die empfohlene Dosis zu erhalten. Die Dosis ist so gleichmässig wie möglich zwischen den beiden Flaschen aufzuteilen (z.B. 6 ml und 7 ml für eine Dosis von 325 mg). Die erste Flasche ist zuzubereiten und die Dosis ist zu verabreichen, bevor die zweite Flasche zubereitet wird.
Die Suspension zum Einnehmen ist unmittelbar nach der Zubereitung gemäss Gebrauchsanweisung mit der mitgelieferten Applikationsspritze für Zubereitungen zum Einnehmen (in der Packung enthalten) in den Mund oder über eine Magensonde (nicht enthalten, mindestens 12 Charrière, Polyurethan; Kompatibilität gezeigt) zu verabreichen.
Wenn die orale Suspension nicht innerhalb von 15 Minuten nach der Zubereitung verabreicht wird, weisen Sie den Patienten an, sie wegzuwerfen. Bei Verzögerung der Verabreichung von mehr als 15 Minuten, könnte es zu Gel-Bildung kommen.
Die Anweisungen für die Rekonstitution des OJEMDA Pulvers zur Herstellung einer Suspension zum Einnehmen sind am Ende dieser Fachinformation und in der Patienteninformation zu finden.
Kontraindikationen
Überempfindlichkeit gegen den Wirkstoff oder einen der in der Rubrik "Zusammensetzung" genannten Hilfsstoffe.
Warnhinweise und Vorsichtsmassnahmen
Intratumorale Blutung
Bei Patienten unter Tovorafenib wurde sehr häufig über intratumorale Blutungsereignisse (einschliesslich der Bezeichnung Tumorblutung und intrakranielle Tumorblutung) berichtet (siehe Rubrik "Unerwünschte Wirkungen" ). Patienten und Betreuungspersonen sollen auf das Risiko intratumoraler Blutungen während der Behandlung mit Tovorafenib hingewiesen werden. Das Risiko von Tumorblutungen kann bei gleichzeitiger Anwendung von Antikoagulanzien und Thrombozytenaggregationshemmern erhöht sein. Eine Überwachung auf Anzeichen und Symptome einer Blutung und die Beurteilung, soweit klinisch indiziert, sollte routinemässig erfolgen. Das Auftreten von Blutungsereignissen sollte durch eine Unterbrechung der Dosierung oder ein Absetzen der Behandlung gehandhabt werden (siehe Rubrik "Dosierung/ Anwendung" ).
Andere Blutungsereignisse
Bei Patienten, die Tovorafenib einnehmen, wurde sehr häufig über Blutungsereignisse berichtet. Wenn eine Blutung auftritt, sind die Patienten nach klinischer Indikation zu behandeln (siehe Rubrik "Unerwünschte Wirkungen" ).
Patienten und Betreuungspersonen sollen auf das Risiko einer Blutung während der Behandlung mit Tovorafenib hingewiesen werden. Das Risiko einer Blutung kann bei gleichzeitiger Anwendung von Antikoagulanzien und Thrombozytenaggregationshemmern erhöht sein. Eine Überwachung auf Anzeichen und Symptome einer Blutung und die Beurteilung, soweit klinisch indiziert, sollte routinemässig erfolgen. Das Auftreten von Blutungsereignissen sollte durch eine Unterbrechung der Dosierung oder ein Absetzen der Behandlung gehandhabt werden (siehe Rubrik "Dosierung/Anwendung" ).
Auswirkung auf das Wachstum
Bei Patienten unter Tovorafenib wurde sehr häufig über eine verringerte Wachstumsrate berichtet (siehe Rubrik "Unerwünschte Wirkungen" ). Patienten und Betreuungspersonen sollen auf das Risiko von Auswirkungen auf das Wachstum während der Behandlung mit Tovorafenib hingewiesen werden. Die Überwachung des Wachstums und der Entwicklung sollte vor dem Beginn, routinemässig während und nach Absetzen der Behandlung mit Tovorafenib erfolgen (siehe Rubrik "Dosierung/Anwendung" ).
Leberbezogene Ereignisse
Bei Patienten unter Tovorafenib wurde sehr häufig über leberbezogene Ereignisse, insbesondere einer Erhöhung der Alaninaminotransferase (ALT), der Aspartataminotransferase (AST) und des Bilirubins, berichtet (siehe Rubrik "Unerwünschte Wirkungen" ).
Die Überwachung von Leberfunktionstests, einschliesslich der AST-, ALT- und Bilirubinwerte, sollte vor dem Beginn, ein Monat nach dem Beginn und routinemässig während der Behandlung mit Tovorafenib erfolgen. Je nach Schweregrad sollte die Behandlung ausgesetzt, nach Verbesserung entweder mit der gleichen oder einer reduzierten Dosierung wieder aufgenommen oder dauerhaft abgesetzt werden (siehe Rubrik "Dosierung/Anwendung" ).
Hauttoxizität, einschliesslich Photosensitivität
Bei Patienten unter Tovorafenib wurde sehr häufig über Ausschlag, einschliesslich Photosensitivitätsereignisse berichtet (siehe Rubrik "Unerwünschte Wirkungen" ). Patienten sollten auf neue oder sich verschlechternde Hautreaktionen überwacht werden. Dermatologische Beratung und der Beginn einer unterstützenden Pflege sollte, soweit klinisch indiziert, in Erwägung gezogen werden. Patienten und Betreuungspersonen sollten auf das Risiko von Ausschlag und Photosensitivität unter der Behandlung mit Tovorafenib hingewiesen werden. Es wird empfohlen während der Behandlung mit Tovorafenib Schutzmassnahmen gegen ultraviolette Strahlung zu ergreifen, etwa die Anwendung eines Sonnenschutzpräparats (LSF ≥50), das Tragen einer Sonnenbrille und/oder von Schutzkleidung. Je nach Schweregrad der unerwünschten Wirkung sollte die Behandlung ausgesetzt, bei reduzierter Dosierung wieder aufgenommen oder dauerhaft abgesetzt werden (siehe Rubrik "Dosierung/Anwendung" ).
Embryo-fetale Toxizität und Frauen im gebärfähigen Alter / Verhütung bei Frauen und Männern
Basierend auf tierexperimentellen Daten und dem Wirkmechanismus kann OJEMDA bei Verabreichung an schwangere Frauen den Fötus schädigen (siehe Rubrik "Schwangerschaft, Stillzeit" und "Präklinische Daten" ). Entsprechend ist bei Frauen im gebärfähigen Alter vor Beginn der Behandlung mit Tovorafenib ein Schwangerschaftstest durchzuführen. Vor der Einleitung der Behandlung bei Frauen im gebärfähigen Alter oder bei männlichen Patienten mit Partnerinnen im gebärfähigen Alter ist eine geeignete Beratung über wirksame inklusive nicht-hormonelle Verhütungsmethoden durchzuführen (siehe Rubrik "Schwangerschaft, Stillzeit" ).
Neurofibromatose Typ 1 (NF1) assoziierte Tumoren
Präklinische Daten in NF1 Modellen ohne BRAF-Alterationen haben gezeigt, dass Tovorafenib das Tumorwachstum bei Patienten mit NF1-assoziierten Tumoren fördern kann (siehe Rubrik "Eigenschaften/Wirkungen" ). Vor Einleitung der Behandlung mit Tovorafenib muss die Evidenz einer BRAF-Alteration bestätigt werden.
Natrium
OJEMDA Filmtabletten enthalten weniger als 1 mmol (23 mg) Natrium pro Filmtablette, d. h., sie sind nahezu "natriumfrei" .
Die OJEMDA Suspension zum Einnehmen enthält weniger als 1 mmol (23 mg) Natrium pro Flasche OJEMDA Pulver zur Herstellung einer Suspension zum Einnehmen, d. h., sie ist nahezu "natriumfrei" .
Interaktionen
In vitro Studien
Wirkungen anderer Arzneimittel auf Tovorafenib
Tovorafenib ist ein Substrat des metabolisierenden Enzyms CYP2C8.
Starke oder moderate CYP2C8-Inhibitoren
Legt man ein mechanistisches Verständnis der Ausscheidung von Tovorafenib zugrunde, werden starke oder moderate CYP2C8-Inhibitoren die Tovorafenib-Exposition voraussichtlich erhöhen, wodurch das Risiko unerwünschter Wirkungen bei Tovorafenib steigen kann (siehe Rubrik "Pharmakokinetik" ). Die gleichzeitige Anwendung von Tovorafenib mit einem starken oder moderaten CYP2C8-Inhibitor soll vermieden werden.
Starke oder moderate CYP2C8-Induktoren
Legt man ein mechanistisches Verständnis der Ausscheidung von Tovorafenib zugrunde, werden starke oder moderate CYP2C8-Induktoren die Tovorafenib-Exposition voraussichtlich verringern, was die Wirksamkeit von Tovorafenib reduzieren kann (siehe Rubrik "Pharmakokinetik" ). Die gleichzeitige Anwendung von Tovorafenib mit einem starken oder moderaten CYP2C8-Induktor soll vermieden werden.
Transportersysteme
Tovorafenib ist kein Substrat vom Brustkrebs-Resistenz-Protein (breast cancer resistance protein, BCRP), P-Glykoprotein (P-gp), OATP1B1 und OATP1B3. Tovorafenib wurde nicht als Substrat von OAT1, OAT3, MATE1, MATE2-K und OCT2 bewertet.
Wirkungen von Tovorafenib auf andere Arzneimittel
CYP3A-Substrate
Tovorafenib ist ein CYP3A-Induktor. Die gleichzeitige Anwendung von Tovorafenib verringert voraussichtlich die Exposition bestimmter CYP3A-Substrate und kann dadurch die Wirksamkeit dieser Substrate reduzieren. Die gleichzeitige Anwendung von Tovorafenib mit bestimmten CYP3A-Substraten, bei denen minimale Änderungen der Konzentration zu schwerwiegendem Therapieversagen führen können, soll vermieden werden. Wenn die gleichzeitige Anwendung unvermeidlich ist, überwachen Sie den Patienten auf Wirksamkeitsverlust, sofern die Produktkennzeichnung für CYP3A-Substrate keine anderen Empfehlungen enthält.
Die gleichzeitige Anwendung von Tovorafenib mit hormonellen Kontrazeptiva (CYP3A-Substrate) kann zur Unwirksamkeit der hormonellen Kontrazeptiva führen (siehe die Rubriken "Schwangerschaft/Stillzeit" und "Pharmakokinetik" ). Die gleichzeitige Anwendung von hormonellen Kontrazeptiva mit Tovorafenib soll vermieden werden. Wenn die gleichzeitige Anwendung unvermeidlich ist, muss während der gleichzeitigen Anwendung und 28 Tage lang nach Absetzen von Tovorafenib eine zusätzliche wirksame nicht-hormonelle Verhütungsmethode angewendet werden.
CYP1A2, CYP2B6, CYP2C8, CYP2C9 und CYP2C19 Substrate
Tovorafenib hemmt CYP2C8, CYP2C9, CYP2C19 und CYP3A, jedoch nicht CYP1A2, CYP2B6 und CYP2D6 bei klinisch relevanten Konzentrationen. Tovorafenib induziert CYP2C8, CYP1A2, CYP2B6, CYP2C9 und CYP2C19 bei klinisch relevanten Konzentrationen. Die klinische Relevanz dieser Befunde ist nicht bekannt. Bei gleichzeitiger Anwendung von Tovorafenib mit Arzneimitteln, welche durch diese Enzyme metabolisiert werden, wird eine angemessene Überwachung empfohlen.
Transportersubstrate
Tovorafenib kann möglicherweise BCRP, OATP1B1, OATP1B3 und MATE1 hemmen. Die klinische Relevanz dieser Befunde ist nicht bekannt. Bei gleichzeitiger Anwendung von Tovorafenib mit Arzneimitteln, die Substrate dieser Transporter sind, wird eine angemessene Überwachung empfohlen.
Modellbasierte Ansätze
CYP3A Substrate: Midazolam (CYP3A4 Substrat) Cmax und AUC im Steady-State werden nach gleichzeitiger Anwendung mit Tovorafenib voraussichtlich um mindestens 20% abnehmen.
Schwangerschaft, Stillzeit
Frauen im gebärfähigen Alter / Verhütung bei Frauen und Männern
Bei Frauen im gebärfähigen Alter ist vor Beginn der Behandlung mit Tovorafenib ein Schwangerschaftstest durchzuführen.
Frauen im gebärfähigen Alter müssen während der Therapie und 28 Tage lang nach Absetzen von Tovorafenib wirksame Verhütungsmethoden anwenden. Tovorafenib kann die Wirksamkeit hormoneller Kontrazeptiva verringern, und es sollte eine wirksame nicht-hormonelle Verhütungsmethode angewendet werden (siehe Rubrik "Interaktionen" ). Männliche Patienten mit Partnerinnen im gebärfähigen Alter müssen während der Behandlung mit Tovorafenib und 2 Wochen lang nach der letzten Dosis Kondome benutzen und wirksame Verhütungsmethoden anwenden.
Schwangerschaft
Bisher liegen keine Daten über die Anwendung von Tovorafenib bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Rubrik "Präklinische Daten" ). Tovorafenib sollte schwangeren Frauen nicht verabreicht werden, es sei denn, der mögliche Nutzen für die Mutter übersteigt das mögliche Risiko für den Fötus.
Stillzeit
Es ist nicht bekannt, ob Tovorafenib in die Muttermilch übergeht. Da ein Risiko für das gestillte Kind nicht ausgeschlossen werden kann, ist das Stillen während der Behandlung mit Tovorafenib und 2 Wochen lang nach der letzten Dosis zu unterbrechen.
Fertilität
Bisher liegen keine Daten über die Auswirkungen von Tovorafenib auf die Fertilität bei Menschen vor. Nach Erkenntnissen aus tierexperimentellen Studien kann Tovorafenib sich auf die Fertilität von Männern und Frauen im gebärfähigen Alter auswirken (siehe Rubrik "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Tovorafenib hat einen geringen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen. Der klinische Status der Patienten und das Nebenwirkungsprofil von Tovorafenib sollten bei der Beurteilung der Fähigkeit des Patienten, Aufgaben auszuführen, die Urteilsvermögen, motorische oder kognitive Fähigkeiten erfordern, berücksichtigt werden. Die Patienten sind darauf hinzuweisen, dass Tovorafenib Müdigkeit verursachen kann, was sich auf diese Tätigkeiten auswirken kann.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Das Sicherheitsprofil von Tovorafenib beruht auf den gepoolten Daten von 137 Patienten im Alter von 6 Monaten oder älter mit rezidiviertem oder refraktärem pädiatrischem LGG mit einer BRAF-Alteration in einer klinischen Studie (FIREFLY-1, Arm 1 und 2). Die Behandlungsdauer betrug im Median 22,5 Monate (Spanne: 0,7 bis 32,1 Monate). Die Sicherheitspopulation umfasste Patienten mit einem medianen Alter von 9 Jahren (Spanne: 1 bis 24 Jahre); 3 Patienten (2%) waren 6 Monate bis < 2 Jahre alt, 93 Patienten (68%) waren 2 bis < 12 Jahre alt und 41 Patienten (30%) waren älter als 12 Jahre.
Zu den häufigsten schwerwiegenden unerwünschten Arzneimittelwirkungen zählten Wachstumsverzögerung (6,6%), Erbrechen (6,6%) und Tumorblutung (5,1%).
Die häufigsten unerwünschten Wirkungen nach individueller MedDRA Bezeichnung waren Änderungen der Haarfarbe (77,4%), Kreatinphosphokinase im Blut erhöht (62,0%), Ermüdung (60,6%), Anämie (60,6%), Erbrechen (56,2%), Kopfschmerzen (52,6%), Hypophosphatämie (52,6%), makulo-papulöses Exanthem (50,4%), Fieber (46,7%), Wachstumsverzögerung (43,1%), trockene Haut (40,9%), Aspartataminotransferase erhöht (38,0%), Laktatdehydrogenase im Blut erhöht (38,0%), Übelkeit (37,2%), Obstipation (36,5%), Infektion der oberen Atemwege (35,8%), akneiforme Dermatitis (34,3%), Epistaxis (32,1%), verminderter Appetit (29,9%), Paronychie (29,9%).
Das häufigste unerwünschte Ereigniss, welches zu einer Dosisreduktion von Tovorafenib bei > 5% der Patienten führte, war Makulo-papulöser Ausschlag (5,1%). Die häufigsten berichteten unerwünschten Ereignisse, welche zu einem Aussetzen der Tovorafenib Dosis bei > 5% der Patienten führten, waren Fieber (13,9%), Makulo-papulöser Ausschlag (10,2%), Erbrechen (10,2%), Ermüdung (5,8%), Übelkeit (5,1%), Kopfschmerzen (5,1%) und Alaninaminotransferase erhöht (5,1%). Unerwünschte Ereignisse, die zu einem dauerhaften Absetzen von Tovorafenib bei mehr als einem Patienten führten, waren Tumorblutung (2,9%) und Wachstumsverzögerung (2,9%).
Liste der unerwünschten Wirkungen
Innerhalb der Systemorganklasse werden die Nebenwirkungen nach Häufigkeit aufgeführt und wie folgt klassifiziert: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1'000, < 1/100), selten (≥1/10'000, < 1/1'000), sehr selten (< 1/10'000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 6: Bei pädiatrischen LGG-Patienten in der klinischen pivotalen Studie genannte unerwünschte Arzneimittelwirkungen (n = 137)
Infektionen und parasitäre Erkrankungen
Sehr häufig Infektion der
oberen Atemwege
(35,8%), Paronychie
(29,9%), Virusinfekt
ion (11,7%)
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig Anämiea (61,3%),
Lymphozytenzahl
erniedrigt (16,8%),
Leukozytenzahl
erniedrigt (11,7%)
Häufig Eosinophilenzahl
erhöht
Stoffwechsel- und Ernährungsstörungen
Sehr häufig Verminderter Appetit
(29,9%), Hypokaliäm
ie (28,5%), Hypoalbu
minämie (14,6%),
Hyponatriämie
(13,9%)
Erkrankungen des Nervensystems
Sehr häufig Kopfschmerzen
(52,6%)
Augenerkrankungen
Häufig Blepharitis, trocken
es Auge
Herzerkrankungen
Häufig Ventrikuläre Arrythm
ieb
Gefässerkrankungen
Sehr häufig Blutungc (40,1%),
intratumorale
Blutungd (13,9%),
Flush (10,2%)
Erkrankungen des Gastrointestinaltrakts
Sehr häufig Erbrechen (56,2%),
Übelkeit (37,2%),
Obstipation (36,5%),
Abdominalschmerze
(29,2%), Stomatitisf
(28,5%), Diarrhög
(26,3%)
Leber- und Gallenerkrankungen
Sehr häufig Alaninaminotransfera
se (ALT) erhöht
(24,8%), Bilirubin
im Blut erhöht
(14,6%)
Erkrankungen der Haut und des Unterhautgewebes
Sehr häufig Ausschlagh (83,2%),
Änderungen der
Haarfarbe (77,4%),
trockene Hauti
(47,4%), akneiforme
Dermatitisj (38,0%),
Pruritus (27,7%),
Hautverfärbungk
(20,4%), Alopezie
(20,4%), Lichtempfin
dlichkeitsreaktion
(14,6%)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig Wachstumsverzögerung
l (44,5%), Schmerz
in einer Extremität
(20,4%), Myalgie
(16,1%), Arthralgie
(13,9%)
Allgemeine Erkrankungen
Sehr häufig Ermüdung (60,6%),
Fieber (46,7%),
Ödemm (33,6%)
Untersuchungen
Sehr häufig Kreatinphosphokinase
(CPK) im Blut
erhöht (62,0%),
Phosphat im Blut
erniedrigtn (55,5%),
Aspartataminotransf
erase (AST) erhöht
(38,0%), Laktatdehyd
rogenase (LDH) im
Blut erhöht (38,0%),
Gewicht erniedrigt
(25,5%)
a Umfasst Hämoglobin erniedrigt b Umfasst ventrikuläre Extrasystolen c
Umfasst Epistaxis, Kontusion, Zahnfleischbluten, Hämatom, Petechien,
gastrointestinale Blutung, Hämatemesis, Hämatochezie, Blutung im unteren
Gastrointestinaltrakt, Purpura, subdurale Blutung, vaginale Blutung d Umfasst
Tumorblutung, intrakranielle Tumorblutung e Umfasst Schmerzen im Oberbauch f
Umfasst Cheilitis, Cheilitis angularis, aphthöses Ulkus, Mundulzeration,
Lippenulzeration g Umfasst Enterokolitis h Umfasst makulo-papulöses Exanthem,
Ekzem, erythematösen Ausschlag, papulösen Ausschlag, pustulösen Ausschlag,
Dermatitis, Medikamentenausschlag, Exfoliation der Haut, bullöse Dermatitis,
follikulären Ausschlag, makulösen Ausschlag, Ausschlag mit Juckreiz, Erythema
multiforme, blasigen Hautausschlag i Umfasst rissige Lippen,
Lippentrockenheit, Xeroderma j Umfasst Akne k Umfasst Hautdepigmentierung,
Hauthyperpigmentierung, Hauthypopigmentierung, melanozytischen Nävus l
Umfasst Wachstumsstörung m Umfasst Gesichtsödem, Periorbitalödem, peripheres
Ödem, periphere Schwellung, Lippenödem, schwellendes Gesicht, Schwellung des
Auges, Vulvaödem n Umfasst Hypophosphatämie
Beschreibung spezifischer unerwünschter Wirkungen
Intratumorale Blutung (intratumoural haemorrhage, ITH)
In der Studie FIREFLY-1 wurde intratumorale Blutung (schliesst die Bezeichnungen Tumorblutung und intrakranielle Tumorblutung mit ein) bei 13,9% der Patienten beobachtet. 3,6% der Patienten berichteten von Grad ≥3 Ereignissen und 0,7% der Patienten von Grad 5 Ereignissen. Bei 2,9% der Patienten wurde Tovorafenib dauerhaft abgesetzt aufgrund von ITH Ereignissen. Ab Beginn der Tovorafenib Behandlung betrug die mittlere Zeit bis zum Auftreten 239,2 Tage (Median: 206 Tage; Spanne 23 bis 671 Tage) und die mittlere Dauer des initialen Auftretens einer ITH 30,8 Tage (Median 19,5 Tage; Spanne: 1 Tag bis 88 Tage).
Andere Blutungsereignisse
In der Studie FIREFLY-1 wurden andere Blutungsereignisse bei 40,1% der pädiatrischen Patienten beobachtet, wobei Ereignisse vom Grad ≥3 bei 2,2% auftraten. Das häufigste Blutungsereignis (Epistaxis) wurde bei 32,1% der Patienten gemeldet. Es handelte sich in der Mehrzahl um Ereignisse vom Grad 1. Ein Patient hatte ein Grad 3 Epistaxis Ereignis. Ab Beginn der Tovorafenib Behandlung betrug die mittlere Zeit bis zum Auftreten 124,5 Tage (Median: 77 Tage; Spanne 4 bis 617 Tage) und die mittlere Dauer des initialen Auftretens einer Blutung 78,1 Tage (Median 9 Tage; Spanne: 1 Tag bis 428 Tage).
Wachstumsverzögerung
Patienten, die bis zu 24 Monate lang mit Tovorafenib behandelt wurden, wiesen Reduktionen der z-Scores der Körpergrösse im Vergleich zu entsprechenden alters- und geschlechtsspezifischen normativen Daten auf, obgleich bei Kindern mit pädiatrischem LGG gegebenenfalls veränderte Wachstumsraten im Vergleich zu Kindern ohne Krebs zu erwarten sind. In der Studie FIREFLY-1 wurde bei 44,5% der Patienten im Alter bis 18 Jahre über Wachstumsverzögerung berichtet. Die Wachstumsverzögerung führte bei 5,1% der Patienten zum Aussetzen der Verabreichung und bei 2,2% der Patienten zu einer Dosisreduktion. Bei den Patienten, bei denen eine Wachstumsverzögerung auftrat und Röntgenaufnahmen der Hand zur Bestimmung des Knochenalters angefertigt wurden, lagen keine Hinweise auf einen vorzeitigen Verschluss der Wachstumsfugen oder ein Fortschreiten des Knochenalters vor. Die Wachstumsverzögerung führte bei 2,9% der Patienten zu einem dauerhaften Absetzen der Behandlung. Bei den nach Unterbrechung der Behandlung mit Tovorafenib beobachteten Patienten kam es zu einem Wiederanstieg der Wachstumsrate und einer Zunahme der z-Scores.
Leberbezogene Ereignisse
In der Studie FIREFLY-1 wurde bei 24,8% der Patienten unter Tovorafenib von erhöhten ALT Werten berichtet. Erhöhte AST Werte traten bei 38% der Patienten unter Tovorafenib auf. Bei 5,8% bzw. 2,9% der Patienten wurden Grad ≥3 Erhöhungen von ALT bzw. AST beobachtet. Zusätzlich wurde von erhöhten Bilirubinspiegeln bei 14,6% der Patienten berichtet. Die mittlere Zeit bis zum Auftreten erhöhter ALT Werte betrug 215,3 Tage (Spanne 1 Tag bis 672 Tage), bis zum Auftreten erhöhter AST Werte 123,4 Tage (Spanne 12 bis 813 Tage) und bis zum Auftreten erhöhter Bilirubinspiegel 79,6 Tage (Spanne: 13 bis 645 Tage). Erhöhte ALT Werte führten bei 5,1% der Patienten zu einem Aussetzen der Dosis und bei 1,5% zu einer Dosisreduktion; erhöhte AST Werte führten bei 2,9% der Patienten zu einem Aussetzen der Dosis und bei 0,7% zu einer Dosisreduktion. Erhöhte Bilirubinspiegel führten bei 0,7% der Patienten zu einem Aussetzen der Dosis und bei keinem Patienten war deswegen eine Dosisreduktion notwendig.
Kreatinphosphokinase im Blut erhöht
In der Studie FIREFLY-1 berichteten 62,0% der Patienten über Ereignisse erhöhter Kreatinphosphokinase im Blut. 12,4% der Patienten berichteten über Grad ≥3 Ereignisse. Alle Ereignisse waren nicht schwerwiegend. Von den Patienten, die über einen Anstieg der CPK berichteten, meldete die Mehrheit (61,2%) einen Anstieg innerhalb der ersten 4 Wochen nach Beginn der Behandlung mit Tovorafenib. Einige Patienten hatten mehrere Episoden. Ein Anstieg der CPK führte bei 3,6% der Patienten zu einem Aussetzen der Dosierung. Die mittlere Zeit bis zum Auftreten seit Beginn der Behandlung mit Tovorafenib betrug 98,5 Tage (Median: 29 Tage; Spanne 4 bis 701 Tage). Die mittlere Dauer des erstmaligen Auftretens des Ereignisses betrug 238,4 Tage (Median: 122 Tage; Spanne: 8 bis 926 Tage).
Anämie
In der Studie FIREFLY-1 wurde bei 61,3% der Patienten von einer Anämie berichtet. 13,1% der Patienten berichteten über Grad ≥3 Anämieereignisse. Die Mehrzahl dieser Patienten (54,8%) berichtete über ein Anämieereignis innerhalb von 60 Tagen nach Beginn der Behandlung mit Tovorafenib. Bei einem Patienten kam es zu einem schwerwiegenden Ereignis. Keiner der Patienten brach die Behandlung aufgrund einer Anämie ab; 2,2% der Patienten berichteten über eine Anämie, die ein Aussetzen der Behandlung oder eine Dosisanpassung erforderte. Die mittlere Zeit bis zum Auftreten ab Beginn der Behandlung mit Tovorafenib betrug 107,4 Tage (Median 57 Tage; Spanne 8 bis 737 Tage). Die mittlere Dauer des ersten Auftretens der Anämie betrug 207,1 Tage (Median 89,5 Tage; Spanne: 1 Tag bis 826 Tage).
Hauttoxizität, einschliesslich Photosensitivität
In der Studie FIREFLY-1 trat Ausschlag bei 83,2% der Patienten auf. Die meisten Ereignisse waren leicht, wobei Ereignisse vom Grad ≥3 bei 12,4% der Patienten berichtet wurden. Ausschlag führte bei 16,1% der Patienten zu einem Aussetzen der Dosis und bei 8,8% zu einer Dosisreduktion. Ein Patient (0,7%) brach die Behandlung aufgrund eines juckenden Ausschlags ab. Die mittlere Zeit bis zum Auftreten des Ausschlags ab Beginn der Behandlung mit Tovorafenib betrug 87,6 Tage (Median: 14,5 Tage; Spanne: 1 Tag bis 617 Tage) und die mittlere Dauer des erstmaligen Auftretens des Ausschlags betrug 103 Tage (Median: 43 Tage; Spanne 1 Tag bis 777 Tage). Photosensitivität trat bei 14,6% der Patienten auf, einschliesslich eines Ereignisses vom Grad 3 bei einem einzelnen Patienten (0,7%), und führte bei einem Patienten (0,7%) zum Aussetzen der Verabreichung.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle einer Überdosierung berichtet. Wenn es zu einer Überdosierung kommt, ist die Behandlung mit Tovorafenib auszusetzen, und der Patient erforderlichenfalls unterstützend zu behandeln und zu überwachen. Da Tovorafenib eine hohe Plasmaproteinbindung aufweist, ist die Hämodialyse bei der Behandlung einer Überdosierung von Tovorafenib wahrscheinlich unwirksam.
Eigenschaften/Wirkungen
ATC-Code
L01EC04
Wirkungsmechanismus
Tovorafenib ist ein Typ-II-RAF-Kinase-Inhibitor der mutierten BRAF-V600E-, der Wildtyp-BRAF- und der Wildtyp-CRAF-Kinase, mit IC50-Werten von 7,1; 10,1 bzw. 0,7 nM.
Tovorafenib zeigte antitumorale Aktivität in Zellkulturen und Xenograft-Tumormodellen mit BRAF-V600E- bzw. V600D-Mutation. Antitumorale Aktivität zeigte Tovorafenib auch in einem Xenograft-Tumormodell mit einer BRAF-Fusion. Die präklinischen Studien an Tumormodellen mit BRAF-Alterationen deuten darauf hin, dass Tovorafenib keine paradoxe Aktivierung des Signalwegs der Mitogen-aktivierten Proteinkinase (MAPK) induziert, wie sie bei Typ I BRAF Inhibitoren zu beobachten ist.
In vitro erhöhte Tovorafenib in klinisch relevanten Konzentrationen die extrazelluläre signalregulierte Kinase (ERK)-Phosphorylierung in Zellen mit Neurofibromatose-Typ-1-Funktionsverlust (NF1-LOF). Dies deutet auf eine Aktivierung und nicht auf eine Inhibition des MAP-Kinase-Signalwegs hin. In einem gentechnisch veränderten NF1-Mausmodell des plexiformen Neurofibroms ohne BRAF-Alteration zeigte Tovorafenib keine antitumorale Aktivität, und bei 2/12 Mäusen (etwa 17%) wurde eine Zunahme des Tumorvolumens festgestellt, die jedoch nicht statistisch signifikant war.
Pharmakodynamik
Expositions-Wirkungs-Beziehungen
Die Tovorafenib-Exposition ist mit einer Reduktion der Grösse-zu-Alter-z-Scores bei pädiatrischen Patienten assoziiert. Das Risiko eines reduzierten Grösse-zu-Alter-z-Scores bleibt während der Behandlung mit Tovorafenib bestehen.
Eine höhere Tovorafenib-Exposition ist mit einem erhöhten Risiko von Hautausschlägen, erhöhten Leberenzymen (AST und ALT) und erhöhter Kreatinphosphokinase assoziiert.
Die Expositions-Wirkungs-Beziehungen für die Gesamtansprechrate auf Basis des RAPNO-LGG (Response Assessment in Pediatric Neuro-Oncology) und des RANO-LGG (Response Assessment in Neuro-Oncology) waren über den Dosisbereich von 290 bis 476 mg/m2 (das 0,76- bis 1,25-Fache der empfohlenen Dosis) nicht klinisch bedeutsam.
Kardiale Elektrophysiologie
Bei der empfohlenen Tovorafenib-Dosis von 380 mg/m2 oral einmal wöchentlich (höchstens 600 mg) wurde keine mittlere Verlängerung der QT-Dauer um mehr als 20 Millisekunden beobachtet.
Klinische Wirksamkeit
Die Wirksamkeit von Tovorafenib wurde bei pädiatrischen Patienten im Alter ab 6 Monaten in einer multizentrischen, offenen, einarmigen klinischen Studie der Phase II (FIREFLY-1 [Arm 1]) evaluiert. Geeignete Patienten (n=76) mussten ein rezidiviertes oder refraktäres pädiatrisches niedriggradiges Gliom (LGG) mit einer aktivierenden BRAF-Alteration aufweisen. Der Befund wurde in lokalen Laboruntersuchungen ermittelt. Die Patienten mussten ausserdem mindestens eine messbare Läsion gemäss RANO-Kriterien von 2010 aufweisen. Alle Patienten hatten mindestens eine vorangegangene systemische Therapielinie erhalten, und bei allen war die Krankheitsprogression röntgenologisch nachgewiesen. Patienten mit Tumoren, die eine oder mehrere zusätzliche aktivierende molekulare Alterationen (z.B. IDH1/2-Mutationen, FGFR-Mutationen) aufwiesen, oder Patienten mit bekannter oder Verdachtsdiagnose einer Neurofibromatose Typ 1 (NF1) wurden ausgeschlossen.
Die Patienten erhielten Tovorafenib in einer Dosis von etwa 420 mg/m2 oral einmal wöchentlich (Bereich: 290 bis 476 mg/m2, das 0,76- bis 1,25-Fache der empfohlenen Dosis), entsprechend der Körperoberfläche, mit einer Höchstdosis von 600 mg bis zur Krankheitsprogression, zum Verlust des klinischen Nutzens oder dem Auftreten von unzumutbarer Toxizität. Obgleich in der Studie FIREFLY-1 (Arm 1) Tovorafenib-Dosen von 290 mg/m2 bis 476 mg/m2 verabreicht wurden, beträgt die empfohlene Tovorafenib-Dosis 380 mg/m2 oral einmal wöchentlich, weil diese Dosis als sicher und wirksam ermittelt wurde für die Behandlung von Patienten im Alter ab 6 Monaten mit rezidiviertem oder refraktärem pädiatrischem LGG, das eine BRAF-Fusion, ein BRAF-Rearrangement oder eine BRAF-V600-Mutation aufweist (siehe Rubrik "Dosierung/Anwendung" ).
Tumorbeurteilungen wurden alle 12 Wochen durchgeführt.
Der primäre Endpunkt war die Gesamtansprechrate (overall response rate, ORR) der Patienten, welche durch unabhängige Prüfung auf Basis der RANO-HGG (Response Assessment in Neuro Oncology for High-Grade Glioma) beurteilt wurde. Die ORR wurde als sekundärer Endpunkt auf Basis der RAPNO-LGG geprüft und als exploratorischer Endpunkt auf Basis der RANO-LGG. Weitere Wirksamkeitsendpunkte wurden auf Basis der drei Response Assessments geprüft.
Das mediane Alter betrug 8,5 Jahre (Spanne: 2 bis 21 Jahre); 14 Patienten waren jünger als 6 Jahre, 42 zwischen 6 und 12 Jahre, 15 zwischen 12 und 16 Jahre und 6 Patienten waren älter als 16 aber jünger als 25 Jahre alt. 53% waren männlich; 61% waren weiss und 93% hatten einen Karnofsky/Lansky-Performance-Status von 80 bis 100. Die Patienten hatten im Median 3 vorangegangene systemische Behandlungsschemata (Spanne: 1 bis 9) erhalten, wobei 22% ein, 26% zwei, 21% drei und 30% mehr als drei vorangegangene systemische Behandlungsschemata erhalten hatten. 46 Patienten (60%) hatten eine vorangegangene Behandlung mit einem MAP-Kinase-Inhibitor erhalten. Die Tumoren waren in den meisten Fällen innerhalb der Sehbahn (51%), der tiefen Mittellinienstrukturen (12%), des Hirnstamms (8%), des Kleinhirns (7%) und einer Grosshirnhemisphäre (5%) lokalisiert. 63 Patienten (83%) wiesen eine BRAF-Fusion oder ein BRAF-Rearrangement und 13 Patienten (17%) eine V600-Mutation auf.
Die mediane Behandlungsdauer betrug 23,7 Monate (Spanne: 0,7 bis 32,1 Monate). Per Protokoll konnten die Patienten auch nach 26 Therapiezyklen/ 24 Monaten und nach Ermessen des Prüfarztes eine optionale Therapiepause einlegen: 43% der Patienten (33/76) machten eine Therapiepause, und 14% der Patienten (11/76) erhielten die Behandlung weiterhin. Von den Patienten, welche eine Therapiepause einlegten, wurden 3 Patienten (9,1%) nach klinischer oder radiographischer Evidenz der Krankheitsprogression mit Tovorafenib wiederbehandelt.
Die Wirksamkeitsergebnisse basierend auf dem 2-Jahres-Nachbeobachtungszeitraum sind in Tabelle 7 zusammengefasst.
Tabelle 7: Wirksamkeitsergebnisse auf Basis der unabhängigen Überprüfung in FIREFLY-1 (Arm 1)
Wirksamkeitsparameter RANO-HGG (primärer Endpunkt) N=69* RAPNO-LGG N = 76* Bestes Gesamtansprechen Vollständiges Ansprechen (CR), n (%) 16 (23,2%) 0 (0) Partielles Ansprechen (PR), n (%) 33 (47,8%) 29 (38,2%) Geringes Ansprechen (MR), n (%) NA 11 (14,5%) Stabile Erkrankung (SD), n (%) 15 (21,7%) 22 (28,9%) Gesamtansprechrate ORR (CR+PR+MR) 95%-KIa 71,0% (58,8; 81,3) 52,6% (40,8; 64,2) Rate des klinischen Nutzens (95% KI)ab 76,8% (65,1; 86,1) 57,9% (46,0; 69,1) Ansprechdauer (DoR) N=49 N = 40 Median (95%-KI)c, Monate 19,7 (13,7; NE) 18,0 (12,0; 22,8) DoR-Rate nach ≥12 Monatenc 66,0% 65% DoR-Rate nach ≥24 Monatenc 43,4% 25,6% Progressionsfreies Überleben (PFS) Median PFS (95% KI) 22,3 (16,5; 25,1) 16,6 (8,3; 17,1)
Abkürzungen: RANO-HGG= Response Assessment in Neuro-Oncology for High-Grade Glioma; RAPNO-LGG = Response Assessment in Pediatric Neuro-Oncology for Low-Grade Glioma; NA= nicht anwendbar; KI = Konfidenzintervall; NE= nicht abschätzbar.
*Mindestens eine messbare Läsion durch das relevante Bildgebungskriterium bei Baseline auf Basis der RAPNO-LGG und RAPNO-HGG-Kriterien.
a Auf Basis des exakten Konfidenzintervalls nach Clopper-Pearson.
b Auf Basis der CR, PR oder der stabilen Erkrankung einer Dauer von ≥12 Monaten für die RANO-HGG Kriterien. Auf Basis der CR, PR, MR oder der stabilen Erkrankung einer Dauer von ≥12 Monaten für die RAPNO-LGG Kriterien.
c Auf Basis der Kaplan-Meier-Schätzung.
Unter den Respondern basierend auf den RANO-HGG Kriterien betrug die mediane Zeit bis zum Ansprechen 5,3 Monate (Spanne: 2,6 bis 19,4). Die ORR betrug 74,6% bei den Patienten mit BRAF-Fusion oder -Rearrangement (n = 59) bzw. 50% bei Patienten mit BRAF-V600E-Mutation (n = 10). Die ORR betrug 73% bei Patienten, die vorher eine gegen die MAPK gerichtete Therapie erhalten hatten (n = 41), und 67,9% bei Patienten, die vorher keine gegen die MAPK gerichtete Therapie erhalten hatten (n = 28).
Unter den Respondern basierend auf den RAPNO-LGG Kriterien betrug die mediane Zeit bis zum Ansprechen 5,4 Monate (Spanne: 1,6 bis 17,5). Die ORR betrug 53% (95%-KI: 40,2; 65,7) bei Patienten mit BRAF-Fusion oder -Rearrangement (n = 64) bzw. 50% (95%-KI: 21,1; 78,9) bei Patienten mit BRAF-V600E-Mutation (n = 12). Die ORR betrug 51% (95%-KI: 35,8; 66,3) bei Patienten, die vorher eine gegen die MAPK gerichtete Therapie erhalten hatten (n = 45), und 55% (95%-KI: 36,0; 72,7) bei Patienten, die vorher keine gegen die MAPK gerichtete Therapie erhalten hatten (n = 31). Die mediane DoR betrug 16,6 Monate (95%-KI 11,3; 19,4) bei Patienten, die vorher eine gegen die MAPK gerichtete Therapie erhalten hatten (n = 45), und 22,8 Monate (95%-KI 8,4; -) bei Patienten, die vorher keine gegen die MAPK gerichtete Therapie erhalten hatten (n = 31).
Auf Basis der RANO-LGG-Kriterien (2011) (n = 76) betrug die ORR 54% (95%-KI: 42; 65), wobei 23 Patienten ein PR und 18 Patienten ein MR aufwiesen. 22 Patienten (29%) hatten eine stabile Erkrankung. Drei Patienten im Alter zwischen 11 Monaten und 2 Jahren wurden mit Tovorafenib in Arm 2 behandelt und hatten als bestes Ansprechen eine stabile Erkrankung, vollständiges Ansprechen, sowie partielles Ansprechen (je n=1) basierend auf RANO-HGG Kriterien evaluiert durch den Prüfarzt.
Pharmakokinetik
Sofern nicht anders angegeben, werden die pharmakokinetischen Parameter von Tovorafenib als Mittelwert (CV%) ausgedrückt. Die Höchstkonzentration von Tovorafenib im Steady-State (Cmax) beträgt 6,9 µg/ml (23%) und die Fläche unter der Konzentrations-Zeit-Kurve (AUC) 508 µg*h/ml (31%). Die Zeit bis zum Erreichen des Steady-State beträgt 12 Tage (33%). Die Tovorafenib-Exposition steigt dosisproportional. Es kommt zu keiner klinisch bedeutsamen Akkumulation von Tovorafenib.
Absorption
Die mediane (minimale; maximale) Zeit bis zum Erreichen der maximalen Plasmakonzentration von Tovorafenib (Tmax) beträgt 3 Stunden (1,5; 4 Stunden) nach einer Einzeldosis Tabletten oder Suspension zum Einnehmen.
Nahrungseinfluss
Es wurden keine klinisch bedeutsamen Unterschiede bei der Cmax und der AUC von Tovorafenib nach Verabreichung von Tabletten mit einer fettreichen Mahlzeit (etwa 859 Gesamtkalorien, 54% Fett) im Vergleich zur Verabreichung im Nüchternzustand festgestellt, aber die Tmax war auf 6,5 Stunden verlängert.
Distribution
Das apparente Verteilungsvolumen von Tovorafenib beträgt 60 l/m2 (23%). Tovorafenib wird in vitro zu 97,5% an humane Plasmaproteine gebunden.
Metabolismus
Tovorafenib wird in vitro hauptsächlich durch Aldehydoxidase und CYP2C8 metabolisiert. CYP3A, CYP2C9 und CYP2C19 metabolisieren Tovorafenib in geringem Umfang.
Elimination
Die terminale Halbwertszeit von Tovorafenib beträgt etwa 56 Stunden (33%), und die apparente Clearance beträgt 0,7 l/h/m2 (31%). Nach einer Einzeldosis von radioaktiv markiertem Tovorafenib fanden sich 66,1% der gesamten radioaktiv markierten Dosis im Stuhl (8,6% unverändert) und 28,7% der Dosis im Urin wieder (0,2% unverändert).
Kinetik spezieller Patientengruppen
Pädiatrische Population
Es wurden keine klinisch bedeutsamen Unterschiede von Tovorafenib aufgrund des Alters beobachtet (Spanne: 1 bis 94 Jahre). Cmax und AUC bei pädiatrischen Patienten im Alter von 11 Monaten bis 17 Jahren lagen im Bereich der Werte, die bei Erwachsenen beobachtet wurden, welche die gleiche Dosis pro Körperoberfläche erhalten hatten.
Nierenfunktionsstörung
Es wurden keine klinisch bedeutsamen Unterschiede von Tovorafenib bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung beobachtet (eGFR ≥30 ml/min/1,73 m2, berechnet mit der Schwartz-Formel oder der MDRD-Formel).
Tovorafenib ist bei Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) nicht untersucht worden.
Leberfunktionsstörung
Bei Patienten mit leichter Leberfunktionsstörung (Bilirubin ≤ obere Normgrenze [upper limit of normal, ULN] und Aspartataminotransferase [AST] > ULN oder Bilirubin > 1x bis 1,5x ULN und beliebiger AST-Wert) wurden keine klinisch bedeutsamen Unterschiede mit Tovorafenib festgestellt.
Tovorafenib ist bei Patienten mit mittelschwerer (Bilirubin > 1,5x bis 3x ULN und beliebiger AST-Wert) oder schwerer (Gesamtbilirubin > 3x ULN und beliebiger AST-Wert) Leberfunktionsstörung nicht untersucht worden (siehe Rubrik "Dosierung/Anwendung" ).
Ethnische Zugehörigkeit
Bezogen auf die ethnische Zugehörigkeit wurden keine klinisch bedeutsamen Unterschiede mit Tovorafenib beobachtet. Bei einer Populations-PK-Analyse wurden keine klinisch relevanten Unterschiede in der PK von Tovorafenib aufgrund unterschiedlicher ethnischer Zugehörigkeit (weiss, schwarz, asiatisch) festgestellt.
Geschlecht
Es wurden keine geschlechtsspezifischen klinisch bedeutsamen Unterschiede in der Pharmakokinetik von Tovorafenib beobachtet.
Präklinische Daten
Mutagenität
Tovorafenib hat sich im bakteriellen In-vitro-Rückmutationstest (Ames-Test) als nicht mutagen erwiesen. Tovorafenib löste in vitro in kultivierten humanen Lymphozyten nur mit metabolischer Aktivierung bei der höchsten Konzentration, die von Präzipitaten begleitet war, Chromosomenaberrationen aus. Tovorafenib erwies sich in einem in-vivo-Mikronukleus-Test an Knochenmark von Ratten als nicht genotoxisch. Somit wird Tovorafenib insgesamt als nicht genotoxisch betrachtet.
Kanzerogenität
Tovorafenib war in einer 26-wöchigen Studie an transgenen Mäusen bei Expositionen, die etwa dem 0,6-Fachen der humanen Exposition (AUC) bei der empfohlenen Humandosis entsprachen, nicht kanzerogen. Eine 2-jährige Kanzerogenitätsstudie in Ratten wird zur Zeit durchgeführt.
Reproduktionstoxizität
In einer Studie zur embryofetalen Entwicklung an trächtigen Ratten wurde bei allen Tieren bei Dosen von ≥37,5 mg/kg/Tag (bezogen auf die AUC etwa das 0,8-Fache der humanen Exposition bei der empfohlenen Dosis) ein Verlust des ganzen Wurfes infolge früher Resorption beobachtet.
In einer Studie zur Fertilität und frühen Embryonalentwicklung bei weiblichen Ratten verringerte Tovorafenib die Zahl der Trächtigkeiten, der Gelbkörper, der Implantationen und der lebenden Embryos und erhöhte die Zahl der Postimplantationsverluste bei Dosen von 37,5 mg/kg/Tag (bezogen auf die AUC etwa das 0,8-Fache der humanen Exposition bei der empfohlenen Dosis).
In toxikologischen Studien mit wiederholten Dosen bei Ratten mit einer Dauer von bis zu 3 Monaten wurden bei weiblichen Ratten unter Tovorafenib in Dosen von ≥50 mg/kg einmal alle zwei Tage (entspricht, bezogen auf die AUC, etwa der 0,4-fachen humanen Exposition bei der empfohlenen Dosis) eine reversible erhöhte Dicke der Vaginalschleimhaut, eine Zunahme der Grösse und/oder der Anzahl der Corpora haemorrhagica und der Blutungen festgestellt, und es wurden nicht reversible zystische Follikel, verringerte Corpora lutea und Hyperplasie der Interstitialzellen in Ovarien beobachtet. Bei männlichen Ratten rief Tovorafenib in Dosen von ≥50 mg/kg einmal alle zwei Tage (entspricht, bezogen auf die AUC, etwa der 0,3-fachen humanen Exposition bei der empfohlenen Dosis) eine Reduktion der Gewichte von Nebenhoden und Hoden hervor; dies korrelierte mit der reversiblen tubulären Degeneration/Atrophie der Hoden und reduzierten epididymalen Spermien.
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Pulver zur Herstellung einer Suspension zum Einnehmen:
Nicht über 25 °C und in der Originalverpackung aufbewahren.
Die Flasche besteht aus Glas. Dieses Arzneimittel darf nicht verwendet werden, wenn die Flasche zerbrochen oder beschädigt ist oder wenn das Sicherheitssiegel unter dem Verschluss gebrochen ist oder fehlt.
Verwenden Sie die rekonstituierte Suspension zum Einnehmen innerhalb von 15 Minuten nach der Zubereitung. Bei Verzögerung der Verabreichung von mehr als 15 Minuten, könnte es zu Gel-Bildung kommen.
Filmtabletten:
Nicht über 25 °C und in der Originalverpackung aufbewahren.
Entnehmen Sie die Tabletten erst unmittelbar vor der Einnahme aus der Blisterpackung.
Zulassungsnummer
70241, 70260 (Swissmedic)
Packungen
Pulver zur Herstellung einer Suspension zum Einnehmen:
1 Flasche (300 mg): transparente Glasflasche mit kindersicherem Schraubverschluss, die zusammen mit einem Flaschen-Steckadapter und einer 20-ml-Applikationsspritze für Zubereitungen zum Einnehmen in einer Packung verpackt ist [A]
Filmtabletten:
16 Filmtabletten: 4 Blisterkarten (zu je 4 Tabletten) pro Packung. [A]
20 Filmtabletten: 4 Blisterkarten (zu je 5 Tabletten) pro Packung. [A]
24 Filmtabletten: 4 Blisterkarten (zu je 6 Tabletten) pro Packung. [A]
Zulassungsinhaberin
IPSEN Pharma Schweiz GmbH, Zug
Stand der Information
Januar 2026
OJEMDA Pulver zur Herstellung einer Suspension zum Einnehmen – Anweisungen für die Rekonstitution
Bitte lesen Sie die Anweisungen für die Rekonstitution jedes Mal aufmerksam durch, bevor Sie eine Dosis OJEMDA zubereiten.
Der Arzt oder Apotheker bzw. die Ärztin oder Apothekerin sollte dem Patienten oder der Betreuungsperson zeigen, wie eine Dosis OJEMDA richtig zubereitet, abgemessen und gegeben wird. Die Rekonstitution kann oral oder über eine nasogastrische Sonde (Polyurethan; Kompatibilität gezeigt) mit einer Mindestgrösse von 12 Charrière unter Verwendung einer ENFIT-Spritze gegeben werden.
OJEMDA Pulver zur Herstellung einer Suspension zum Einnehmen ist wie folgt zu rekonstituieren:
Hinweis: Wenn Sie mehr als eine Flasche rekonstituieren müssen, rekonstituieren Sie bitte eine Flasche nach der anderen.
Der Vorgang ist auf einer sauberen, ebenen Arbeitsfläche mit sauberen Händen durchzuführen.
Schritt 1: Füllen Sie einen Becher zur Hälfte mit Wasser, das Raumtemperatur hat. Kein kaltes Wasser verwenden.
Schritt 2: Ziehen Sie den Kolben der Applikationsspritze für Zubereitungen zum Einnehmen hoch, um Wasser bis zur 14-ml-Marke aufzuziehen.
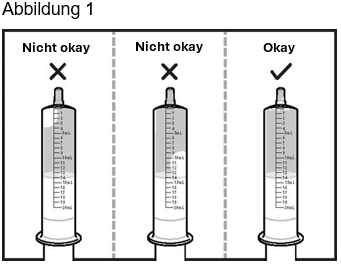
Schritt 3: Drehen Sie die Applikationsspritze für Zubereitungen zum Einnehmen mit der Spitze nach oben und prüfen Sie auf Luftblasen. Wenn in der Applikationsspritze für Zubereitungen zum Einnehmen grosse Luftblasen enthalten sind, drücken Sie das Wasser wieder in den Becher und ziehen Sie das Wasser anschliessend erneut bis zur 14-ml-Marke auf. Wiederholen Sie diesen Schritt, bis keine grossen Luftblasen mehr vorhanden sind. Kleine Luftblasen sind in Ordnung (siehe Abbildung 1).
Schritt 4: Öffnen Sie die Flasche mit dem Pulver. Drücken Sie dazu fest auf den Verschluss und drehen Sie ihn dabei nach links (gegen den Uhrzeigersinn). Das Produkt darf nicht verwendet werden, wenn die Flasche zerbrochen oder beschädigt ist oder wenn das Sicherheitssiegel unter dem Verschluss beschädigt ist oder fehlt. Werfen Sie den Verschluss nicht weg.
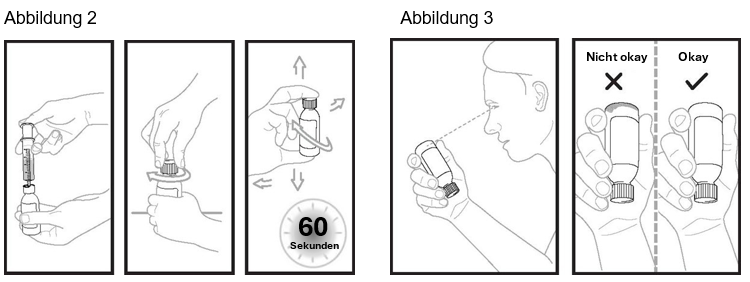
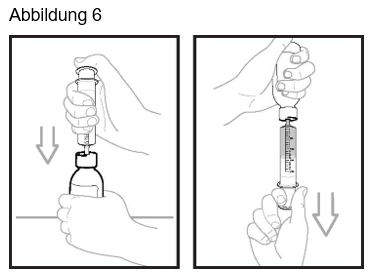
Schritt 5: Spritzen Sie mit der Applikationsspritze für Zubereitungen zum Einnehmen exakt 14 ml Wasser in die Flasche (siehe Abbildung 2). Verschliessen Sie die Flasche sofort wieder. Drücken Sie dazu auf den Verschluss und drehen Sie ihn gleichzeitig nach rechts (im Uhrzeigersinn). Schütteln Sie die Flasche 60 Sekunden lang gut in alle Richtungen.
Drehen Sie die Flasche auf den Kopf, um zu prüfen, ob Pulver an der Flascheninnenseite festklebt (siehe Abbildung 3). Wenn Sie noch Pulver in der Flasche sehen, schütteln Sie die Flasche weitere 15 Sekunden lang, bis Sie in der Flasche kein Pulver mehr sehen. Die Flasche insgesamt nicht länger als 2 Minuten schütteln. Wenn Sie noch Pulver in der Flasche sehen, bitten Sie um eine neue Flasche.
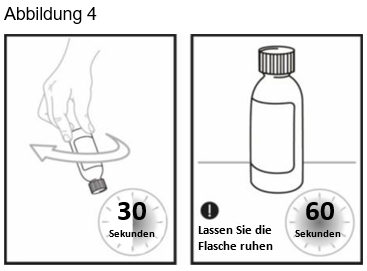
Schritt 6: Drehen Sie Flasche erneut auf den Kopf und schwenken Sie sie 30 Sekunden lang (siehe Abbildung 4). Entfernen Sie den Verschluss und kontrollieren Sie, dass keine Feststoffe im Flaschenhals kleben. Wenn Sie beim Entfernen des Verschlusses Feststoffe im Flaschenhals sehen, verschliessen Sie die Flasche wieder, drehen Sie die Flasche auf den Kopf und schwenken Sie sie weitere 15 Sekunden lang.
Lassen Sie die Flasche 60 Sekunden stehen, damit der Schaum grösstenteils zusammenfallen kann. Hinweis: Die Schaumbildung in der Flasche reduziert die Menge der OJEMDA Suspension zum Einnehmen.
Schritt 7: Drücken Sie den Flaschenadapter fest oben in die Flasche. Die Oberkante des Flaschenadapters sollte bündig mit der Oberseite der Flasche sein.
Entfernen Sie den Flaschenadapter nach dem Einsetzen in die Flasche nicht mehr.
Schritt 8: Prüfen Sie die verschriebene Dosis in Millilitern (ml). Ziehen Sie Luft in die Applikationsspritze für Zubereitungen zum Einnehmen. Ziehen Sie dazu den Kolben bis zur verschriebenen Dosis heraus.
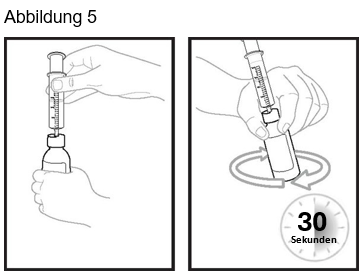
Schritt 9: Führen Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen in den Flaschenadapter ein. Die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen sollte genau in das Loch des Flaschenadapters passen. Halten Sie die Flasche mit der eingesteckten Applikationsspritze für Zubereitungen zum Einnehmen dort fest, wo die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen in den Flaschenadapter eingesetzt ist, und schwenken die Suspension zum Einnehmen 30 Sekunden lang (siehe Abbildung 5).
Schritt 10: Drücken Sie Luft aus der Applikationsspritze für Zubereitungen zum Einnehmen in die Flasche (siehe Abbildung 6). Halten Sie die Applikationsspritze für Zubereitungen zum Einnehmen an Ort und Stelle und drehen Sie die Flasche auf den Kopf. Zur Abmessung der verschriebenen Dosis halten Sie die Applikationsspritze für Zubereitungen zum Einnehmen so, dass ihre Spitze nach oben zeigt, und ziehen Sie den Kolben herunter, bis die Oberseite des Kolbens auf einer Linie mit der verschriebenen Dosis in Millilitern ist.
Schritt 11: Während die Spritze noch im Adapter in der Flasche steckt, entfernen Sie eventuelle Luftblasen aus der Applikationsspritze für Zubereitungen zum Einnehmen. Drücken Sie die OJEMDA Suspension dazu vorsichtig zurück in die Flasche und ziehen dann erneut den Kolben herunter, um Ihre verordnete Dosis aufzuziehen.
Wiederholen Sie diesen Schritt, bis Sie sehen, dass nur wenige oder gar keine Luftblasen verblieben sind, oder falls Sie die falsche Dosis in die Applikationsspritze für Zubereitungen zum Einnehmen aufgezogen haben. Verwenden Sie nur höchstens 12 ml OJEMDA aus jeder zubereiteten Flasche.
Schritt 12: Lassen Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen im Flaschenadapter und drehen Sie die Flasche vorsichtig in die aufrechte Position. Setzen Sie die Flasche wieder auf eine ebene Arbeitsfläche. Ziehen Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen vorsichtig gerade nach oben und entfernen Sie sie langsam aus dem Flaschenadapter. OJEMDA ist bereit zur Anwendung.