Zusammensetzung
Wirkstoffe
Efgartigimod alfa.
Efgartigimod alfa ist ein Fc-Fragment des humanen rekombinanten Immunglobulins G1 (IgG1), das durch rekombinante DNA-Technologie in Ovarialzellen des chinesischen Hamsters (CHO-Zellen) hergestellt wird.
Hilfsstoffe – Vyvgart, Injektionslösung
Rekombinante humane Hyaluronidase (rHuPH20) (gentechnologisch hergestellt unter Verwendung von CHO-Zellen), Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 20 (E 432), Natriumchlorid, Saccharose, Wasser für Injektionszwecke.
Jede Durchstechflasche enthält 12,9 mg Natrium.
Hilfsstoffe - Vyvgart, Injektionslösung in einer Fertigspritze
Rekombinante humane Hyaluronidase (rHuPH20) (gentechnologisch hergestellt unter Verwendung von CHO-Zellen), Argininhydrochlorid, Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 80 (E 433), Natriumchlorid, Saccharose, Wasser für Injektionszwecke.
Jede Fertigspritze enthält 8,1 mg Natrium.
Darreichungsform und Wirkstoffmenge pro Einheit
Injektionslösung.
Jede Durchstechflasche enthält 1000 mg Efgartigimod alfa in 5,6 ml (180 mg/ml).
Jede Fertigspritze enthält 1000 mg Efgartigimod alfa in 5 ml (200 mg/ml).
Gelblich, klar bis opaleszierend, pH 6,0.
Indikationen/Anwendungsmöglichkeiten
Vyvgart wird
zusätzlich zur Standardtherapie zur Behandlung von erwachsenen Patienten mit generalisierter Myasthenia gravis (gMG) angewendet, die Anti-Acetylcholin-Rezeptor (AChR)-Antikörper positiv sind.
als Monotherapie zur Behandlung von erwachsenen Patienten mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen angewendet.
Dosierung/Anwendung
Die Behandlung muss von einem Arzt, der Erfahrung in der Behandlung von Patienten mit neuromuskulären Erkrankungen hat, eingeleitet und überwacht werden.
Übliche Dosierung
Generalisierte Myasthenia gravis
Der erste Behandlungszyklus und die erste Anwendung des zweiten Behandlungszyklus müssen entweder von einem Arzt oder unter dessen Aufsicht durchgeführt werden. Die nachfolgende Behandlung sollte von einem Arzt durchgeführt werden oder kann nach ausreichender Schulung in der subkutanen Injektionstechnik von einem Patienten oder einer Pflegeperson zu Hause durchgeführt werden.
Die empfohlene Dosis beträgt 1000 mg subkutan als einmal wöchentliche Injektion über 4 Wochen (1 Zyklus). Nachfolgende Behandlungszyklen sind der klinischen Beurteilung entsprechend durchzuführen. Die Häufigkeit der Behandlungszyklen kann je nach Patient variieren (siehe Rubrik "Eigenschaften/Wirkungen" ).
Im klinischen Entwicklungsprogramm wurden nachfolgende Behandlungszyklen frühestens 7 Wochen nach der ersten Infusion des vorherigen Zyklus durchgeführt.
Bei Patienten, die derzeit Efgartigimod alfa intravenös erhalten, kann alternativ die Lösung zur subkutanen Injektion verwendet werden. Es wird empfohlen, zu Beginn eines neuen Behandlungszyklus auf die andere Darreichungsform umzustellen. Es liegen keine Daten zur Sicherheit und Wirksamkeit bei Patienten vor, bei denen während desselben Zyklus die Darreichungsform gewechselt wird.
Zur Wirksamkeit bei Patienten mit generalisierter Myasthenia gravis, die zuvor auf eine Plasmapherese-Behandlung (engl. Plasma exchange (PLEX)) nicht angesprochen haben, liegen keine Erfahrungen vor.
Chronisch-entzündliche demyelinisierende Polyneuropathie
Die ersten 4 Injektionen müssen entweder von einem Arzt oder unter dessen Aufsicht durchgeführt werden. Die nachfolgenden Injektionen sollten von einem Arzt durchgeführt werden oder können nach ausreichender Schulung in der subkutanen Injektionstechnik von einem Patienten oder einer Pflegeperson zu Hause durchgeführt werden.
Die empfohlene Dosis beträgt 1000 mg subkutan als einmal wöchentliche Injektion.
Die Behandlung wird mit einem wöchentlichen Dosierungsschema eingeleitet und kann je nach klinischer Beurteilung auf alle zwei Wochen angepasst werden. Im Falle einer Verschlechterung der Symptome sollte die Anwendung einer einmal wöchentlichen Injektion weitergeführt werden.
Bei Patienten, die von ihren derzeitigen CIDP-Therapien umgestellt werden, sollte die Behandlung mit Vyvgart vorzugsweise eingeleitet werden, bevor die klinische Wirkung dieser vorherigen Therapien nachzulassen beginnt.
Ein klinisches Ansprechen wird normalerweise innerhalb von 3 Monaten nach Einleitung der Behandlung mit Efgartigimod alfa subkutan erzielt. Zur Bewertung der Behandlungswirkung sollte 3 bis 6 Monate nach Behandlungsbeginn und danach in regelmässigen Abständen eine klinische Beurteilung in Betracht gezogen werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Es liegen keine Daten bei Patienten mit Leberfunktionsstörung vor. Bei Patienten mit Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Rubrik "Pharmakokinetik" ).
Patienten mit Nierenfunktionsstörungen
Es liegen begrenzte Daten zur Sicherheit und Wirksamkeit bei Patienten mit leichter Nierenfunktionsstörung vor, bei Patienten mit leichter Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit mässiger oder schwerer Nierenfunktionsstörung sind die Daten zur Sicherheit und Wirksamkeit sehr begrenzt (siehe Rubrik "Pharmakokinetik" ).
Ältere Patienten
Bei Patienten ab 65 Jahren ist keine Dosisanpassung erforderlich (siehe Rubrik "Pharmakokinetik" ).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Efgartigimod alfa bei Kindern und Jugendlichen ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Verspätete Dosisgabe
Zwischen zwei aufeinanderfolgenden Anwendungen sollte ein Abstand von mindestens 3 Tagen eingehalten werden. Wenn Anwendungen nicht zum vorgesehenen Zeitpunkt erfolgen können, sollten sie so bald wie möglich und mindestens 3 Tage vor der nächsten Anwendung erfolgen. Steht die nächste Anwendung in weniger als 3 Tagen an, sollte die versäumte Dosis ausgelassen werden, und die nächste Dosis sollte zum vorgesehenen Zeitpunkt angewendet werden.
Art der Anwendung – Vyvgart, Injektionslösung
Dieses Arzneimittel darf nur als subkutane Injektion angewendet werden. Die Injektionslösung darf nicht intravenös verabreicht werden.
Nach der Entnahme der Durchstechflasche aus dem Kühlschrank muss vor der Injektion mindestens 15 Minuten gewartet werden, damit die Lösung Raumtemperatur annehmen kann. Bei der Zubereitung und Verabreichung der Lösung ist eine aseptische Technik anzuwenden. Die Durchstechflasche nicht schütteln (s. Abschnitt "Hinweise für die Handhabung" ).
Während der ersten Anwendungen von Efgartigimod alfa (siehe Rubrik "Dosierung/Anwendung" ) muss eine geeignete Behandlung für injektions- und überempfindlichkeitsbedingte Reaktionen verfügbar sein (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ). Die empfohlenen Injektionsstellen (Abdomen) sollten bei jeder Injektion gewechselt werden, und die Injektion sollte niemals in Muttermale, Narben oder in Bereiche erfolgen, in denen die Haut empfindlich, verletzt, gerötet oder verhärtet ist. Das Volumen von 5,6 ml sollte über einen Zeitraum von 30 bis 90 Sekunden injiziert werden. Sollte der Patient Unbehagen verspüren, kann die Injektion langsamer durchgeführt werden.
Die erste Selbstinjektion muss immer unter Aufsicht eines Arztes durchgeführt werden. Nach ausreichender Schulung in die subkutane Injektionstechnik können Patienten oder Pflegepersonen das Arzneimittel zu Hause selbst injizieren, wenn ein Arzt dies für angemessen hält. Patienten oder Pflegepersonen sind anzuweisen, bei der Injektion von Vyvgart die Anweisungen in der Packungsbeilage zu befolgen.
Ausführliche Hinweise zur Injektion des Arzneimittels sind den Hinweisen für die Handhabung in der Packungsbeilage zu entnehmen.
Art der Anwendung – Vyvgart, Injektionslösung in einer Fertigspritze
Dieses Arzneimittel darf nur als subkutane Injektion angewendet werden. Nicht intravenös anwenden.
Nach der Entnahme der Fertigspritze aus dem Kühlschrank muss vor der Injektion mindestens 30 Minuten gewartet werden, damit die Lösung Raumtemperatur annehmen kann. Eine Sicherheitskanüle, die nicht in der Packung enthalten ist, ist an die Fertigspritze anzuschliessen. Bei der Handhabung der Fertigspritze und während der Anwendung ist eine aseptische Technik anzuwenden. Die Fertigspritze nicht schütteln (s. Abschnitt "Hinweise für die Handhabung" ).
Während der ersten Anwendungen von Efgartigimod alfa (siehe Rubrik "Dosierung/Anwendung" ) muss eine geeignete Behandlung für injektions- und überempfindlichkeitsbedingte Reaktionen verfügbar sein (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ). Die empfohlenen Injektionsstellen (Abdomen) sollten bei jeder Injektion gewechselt werden, und die Injektion sollte niemals in Muttermale, Narben oder in Bereiche erfolgen, in denen die Haut empfindlich, verletzt, gerötet oder verhärtet ist. Das Arzneimittel sollte über einen Zeitraum von etwa 20 bis 30 Sekunden injiziert werden. Sollte der Patient Unbehagen verspüren, kann die Injektion langsamer durchgeführt werden.
Die erste Selbstinjektion muss immer unter Aufsicht eines Arztes durchgeführt werden. Nach ausreichender Schulung in die subkutane Injektionstechnik können Patienten oder Pflegepersonen das Arzneimittel zu Hause selbst injizieren, wenn ein Arzt dies für angemessen hält. Patienten oder Pflegepersonen sind anzuweisen, bei der Injektion von Vyvgart die Anweisungen in der Packungsbeilage zu befolgen.
Ausführliche Hinweise zur Injektion des Arzneimittels sind den Hinweisen für die Handhabung in der Packungsbeilage zu entnehmen.
Kontraindikationen
Überempfindlichkeit gegen den Wirkstoff oder einen der Hilfsstoffe (siehe Rubrik "Zusammensetzung" ).
Warnhinweise und Vorsichtsmassnahmen
Patienten der Klasse V gemäss der Myasthenia Gravis Foundation of America (MGFA)
Die Behandlung von Patienten der MGFA-Klasse V (d.h. myasthene Krise), definiert als Intubation mit oder ohne mechanische Beatmung, ausser im Rahmen der routinemässigen postoperativen Versorgung, mit Efgartigimod alfa wurde nicht untersucht. Es sind die Reihenfolge der Einleitung etablierter Therapien zur Behandlung der MG-Krise und der Gabe von Efgartigimod alfa sowie deren potenzielle Wechselwirkungen zu berücksichtigen (siehe Rubrik "Interaktionen" ).
Infektionen
Da Efgartigimod alfa eine vorübergehende Verringerung des IgG-Spiegels verursacht, kann sich das Infektionsrisiko erhöhen (siehe Rubrik "Unerwünschte Wirkungen" und Rubrik "Eigenschaften/Wirkungen" ). Die häufigsten in klinischen Studien beobachteten Infektionen waren Infektionen der oberen Atemwege und Harnwegsinfektionen (siehe Rubrik "Unerwünschte Wirkungen" ). Patienten sollten während der Behandlung mit Vyvgart auf klinische Anzeichen und Symptome von Infektionen überwacht werden. Bei Patienten mit einer aktiven Infektion sollte das Nutzen-Risiko-Verhältnis einer Fortsetzung oder Unterbrechung der Behandlung mit Efgartigimod alfa bis zum Abklingen der Infektion berücksichtigt werden. Beim Auftreten schwerwiegender Infektionen sollte in Betracht gezogen werden, die Behandlung mit Efgartigimod alfa zu verschieben, bis die Infektion abgeklungen ist.
Injektionsreaktionen und Überempfindlichkeitsreaktionen
In den klinischen Studien wurden Injektionsreaktionen wie Hautausschlag oder Pruritus berichtet (siehe Rubrik "Unerwünschte Wirkungen" ). Diese waren leicht bis mittelschwer. Nach der Markteinführung wurden Fälle von anaphylaktischen Reaktionen bei intravenöser Anwendung von Efgartigimod alfa gemeldet. Die ersten Anwendungen von Vyvgart müssen unter Aufsicht eines Arztes durchgeführt werden (siehe Rubrik "Dosierung/Anwendung" ). Die Patienten sollten nach der Anwendung für 30 Minuten auf klinische Anzeichen und Symptome von Injektionsreaktionen überwacht werden. Im Falle des Auftretens einer Reaktion sollten je nach Schweregrad der Reaktion geeignete unterstützende Massnahmen eingeleitet werden. Nachfolgende Injektionen können auf Grundlage der klinischen Bewertung vorsichtig durchgeführt werden.
Bei Verdacht auf eine anaphylaktische Reaktion ist die Verabreichung von Vyvgart sofort abzubrechen und eine angemessene medizinische Behandlung einzuleiten. Patienten müssen über das mögliche Auftreten und die Anzeichen und Symptome von Überempfindlichkeitsreaktionen und anaphylaktischen Reaktionen informiert und darauf hingewiesen werden, dass sie sich beim Auftreten solcher Reaktionen unverzüglich an ihren Arzt wenden sollten.
Immunisierungen
Alle Impfstoffe sind gemäss den Immunisierungsrichtlinien anzuwenden.
Die Sicherheit der Immunisierung mit Lebendimpfstoffen oder attenuierten Lebendimpfstoffen und die Reaktion auf die Immunisierung mit diesen Impfstoffen während der Behandlung mit Efgartigimod alfa sind nicht bekannt. Bei Patienten, die mit Efgartigimod alfa behandelt werden, wird im Allgemeinen eine Impfung mit Lebendimpfstoffen oder attenuierten Lebendimpfstoffen nicht empfohlen. Wenn eine Impfung mit Lebendimpfstoffen oder attenuierten Lebendimpfstoffen erforderlich ist, sollten diese Impfstoffe mindestens 4 Wochen vor der nächsten Behandlung und mindestens 2 Wochen nach der letzten Dosis Efgartigimod alfa gegeben werden.
Andere Impfstoffe können nach Bedarf zu jedem Zeitpunkt während der Behandlung mit Efgartigimod alfa angewendet werden.
Immunogenität
In der aktiv kontrollierten Studie ARGX-113-2001 wurden bei 12/110 Patienten (11 %) mit gMG vorbestehende Antikörper nachgewiesen, die an Efgartigimod alfa binden. Bei 19/55 Patienten (35 %), die mit Efgartigimod alfa subkutan behandelt wurden, wurden Antikörper gegen Efgartigimod alfa nachgewiesen, verglichen mit 11/55 Patienten (20 %), welche die intravenöse Darreichungsform erhielten. Neutralisierende Antikörper wurden bei 2 (4 %) der mit Efgartigimod alfa subkutan behandelten Patienten nachgewiesen, und bei 2 Patienten (4 %), welche Efgartigimod alfa intravenös erhielten.
In der Studie ARGX-113-1802 wurden bei 13/317 Patienten (4,1 %) mit CIDP vorbestehende Antikörper nachgewiesen, die an Efgartigimod alfa binden. Bei 20/317 der im offenen Teil der Studie (Phase A) behandelten Patienten (6,3 %) und bei 2/111 der im placebokontrollierten Teil (Phase B) behandelten Patienten (1,8 %) wurden Antikörper gegen Efgartigimod alfa nachgewiesen. Neutralisierende Antikörper wurden nur bei 1 Patienten (0,3 %) im offenen Teil der Studie nachgewiesen (siehe Rubrik "Eigenschaften/Wirkungen" ).
In Anbetracht der niedrigen Inzidenz neutralisierender Antikörper kann die Auswirkung von Antikörpern gegen Efgartigimod alfa auf die klinische Wirksamkeit oder Sicherheit, Pharmakokinetik und Pharmakodynamik nicht beurteilt werden.
Therapien mit Immunsuppressiva und Cholinesteraseinhibitoren
Wenn nichtsteroidale Immunsuppressiva, Kortikosteroide und Cholinesteraseinhibitoren reduziert oder abgesetzt werden, sind die Patienten engmaschig auf Anzeichen einer Krankheitsverschlechterung zu überwachen.
Natriumgehalt
Vyvgart, Injektionslösung / Vyvgart, Injektionslösung in einer Fertigspritze enthält weniger als 1 mmol (23 mg) Natrium pro Durchstechflasche / Fertigspritze, d.h. es ist nahezu "natriumfrei" .
Interaktionen
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Efgartigimod alfa kann die Konzentrationen von Substanzen verringern, die an den humanen neonatalen Fc-Rezeptor (FcRn) binden, d.h. von Immunglobulin-Arzneimitteln, monoklonalen Antikörpern oder Antikörper-Derivaten, welche die humane Fc-Domäne der IgG-Subklasse enthalten. Sofern möglich, wird empfohlen, den Beginn der Behandlung mit diesen Arzneimitteln gegebenenfalls bis auf 2 Wochen nach der letzten Dosis Vyvgart zu verschieben. Als Vorsichtsmassnahme sollten Patienten, die Vyvgart erhalten, während sie mit diesen Arzneimitteln behandelt werden, engmaschig auf das beabsichtigte Ansprechen auf diese Arzneimittel überwacht werden.
Plasmaaustausch, Immunadsorption und Plasmapherese können die Konzentration von Efgartigimod alfa im Blut verringern.
Die potenzielle Wechselwirkung mit Impfstoffen wurde in einem nichtklinischen Modell unter Verwendung von Keyhole Limpet Hemocyanin (KLH) als Antigen untersucht. Bei wöchentlicher Verabreichung von 100 mg/kg an Affen ergaben sich keine Auswirkungen auf die Immunantwort nach Immunisierung mit KLH (für weitere Informationen zu Impfungen siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Schwangerschaft, Stillzeit
Schwangerschaft
Es liegen keine Daten zur Anwendung von Efgartigimod alfa während der Schwangerschaft vor. Es ist bekannt, dass Antikörper, einschliesslich therapeutischer monoklonaler Antikörper, aktiv durch die Plazenta transportiert werden (nach 30 Schwangerschaftswochen), indem sie an FcRn binden.
Efgartigimod alfa kann von der Mutter auf den sich entwickelnden Fötus übertragen werden. Da davon auszugehen ist, dass Efgartigimod alfa die mütterlichen Antikörperspiegel senkt und ausserdem die Übertragung mütterlicher Antikörper auf den Fötus hemmt, ist eine Verringerung des passiven Schutzes des Neugeborenen zu erwarten. Daher sind Risiken und Nutzen der Gabe von lebenden/lebend-attenuierten Impfstoffen an Säuglinge, die Efgartigimod alfa in utero ausgesetzt waren, abzuwägen (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Die Behandlung schwangerer Frauen mit Vyvgart sollte nur in Erwägung gezogen werden, wenn der klinische Nutzen die Risiken überwiegt.
Stillzeit
Es liegen keine Informationen über das Vorhandensein von Efgartigimod alfa in der Muttermilch, über Auswirkungen auf das gestillte Kind oder über Auswirkungen auf die Milchproduktion vor. Es wurden keine tierexperimentellen Studien zum Übergang von Efgartigimod alfa in die Milch durchgeführt, daher kann eine Ausscheidung in die Muttermilch nicht ausgeschlossen werden. Es ist bekannt, dass maternales IgG in der Muttermilch vorhanden ist. Es liegen keine Untersuchungen zu möglichen Veränderungen des maternalen IgGs in der Muttermilch und des passiven Schutzes des Neugeborenen unter einer Behandlung mit Vyvgart vor. Die Behandlung stillender Frauen mit Efgartigimod alfa sollte nur in Erwägung gezogen werden, wenn der klinische Nutzen die Risiken überwiegt.
Fertilität
Es liegen keine Daten zur Wirkung von Efgartigimod alfa auf die Fertilität beim Menschen vor. Tierexperimentelle Studien liessen keinen Einfluss von Efgartigimod alfa auf männliche und weibliche Fertilitätsparameter erkennen (siehe Rubrik "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Vyvgart hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die am häufigsten beobachteten unerwünschten Wirkungen waren Reaktionen an der Injektionsstelle (38,2 %, nur subkutan), Kopfschmerzen (12,7%), Infektionen der oberen Atemwege (10,7 %) und Harnwegsinfektionen (9,5 %).
Die Gesamtheit der Sicherheitsdaten deutet darauf hin, dass das Sicherheitsprofil bei beiden Darreichungsformen übereinstimmt (für sowohl zyklische als auch kontinuierliche Dosierungsschemata), mit Ausnahme von Reaktionen an der Injektionsstelle bei Vyvgart (subkutan), die auf den Verabreichungsweg zurückzuführen sind.
Liste der unerwünschten Wirkungen
Die Sicherheit von Vyvgart (intravenös) wurde bei 167 Patienten (84 Patienten behandelt mit Efgartigimod alfa und 83 Patienten behandelt mit Placebo) mit gMG in der 26-wöchigen doppelblinden, placebokontrollierten klinischen Phase-III-Studie (ARGX-113-1704) untersucht.
Weitere Daten zur Langzeitsicherheit wurden an 145 Patienten mit generalisierter Myasthenie erhoben, die im Anschluss an ARGX-113-1704 mit bis zu 19 Zyklen Vyvgart intravenös in der offenen Phase-III-Verlängerungsstudie ARGX-113-1705 behandelt wurden.
Darüber hinaus wurde die Sicherheit von Vyvgart subkutan und intravenös in einer 10-wöchigen offenen, randomisierten, klinischen Phase-III-Parallelgruppenstudie (ARGX-113-2001) bei 110 Patienten mit gMG vergleichend untersucht: 55 Patienten erhielten die intravenöse und 55 Patienten die subkutane Darreichungsform. Die in diesem Abschnitt beschriebenen unerwünschten Wirkungen basieren auf klinischen Studien und auf Berichten nach der Markteinführung.
In Tabelle 1 sind unerwünschte Wirkungen nach Systemorganklasse und bevorzugtem Begriff aufgeführt. Häufigkeitskategorien sind folgenderweise definiert: sehr häufig (≥1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1'000 bis <1/100), selten (≥1/10'000 bis <1/1'000) oder nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden). Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen nach abnehmender Häufigkeit geordnet.
Tabelle 1. Unerwünschte Wirkungen
Systemorganklasse Unerwünschte Wirkung Häufigkeitskategorie
Infektionen und parasitäre Erkrankungen* Infektionen der oberen Atemwege Sehr häufig
(10,7 %)
Harnwegsinfektionen Häufig
Bronchitis Häufig
Erkrankungen des Immunsystems Hautausschlag Häufig
Infusionsreaktionc Häufig
Anaphylaktische Reaktiona Nicht bekannt
Erkrankungen des Nervensystems Kopfschmerzen (12,7 %) Sehr häufig
Erkrankungen des Gastrointestinaltrakts Übelkeit Häufig
Skelettmuskulatur-, Bindegewebs- und Myalgie Häufig
Knochenerkrankungen
Allgemeine Erkrankungen und Beschwerden am Reaktionen an der Injektionsstell Sehr häufig
Verabreichungsort e (38,2 %)b
Fatigue Häufig
Verletzung, Vergiftung und durch Eingriffe Kopfschmerz im Zusammenhang mit Häufig
bedingte Komplikationen* dem Verfahrenc
* Siehe Abschnitt "Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen"
a Von Spontanmeldungen nach der Markteinführung bei intravenöser Anwendung
b Nur bei subkutaner Anwendung (siehe auch Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen)
c Nur bei intravenöser Anwendung.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Reaktionen an der Injektionsstelle (nur bei subkutaner Anwendung)
In der offenen Phase-III-Studie zu gMG (ARGX-113-2001) wurden insgesamt 55 Probanden mit subkutanem Efgartigimod alfa behandelt. Alle Reaktionen an der Injektionsstelle (z.B. Hautausschlag an der Injektionsstelle, Erythem an der Injektionsstelle, Juckreiz an der Injektionsstelle, Schmerzen an der Injektionsstelle) waren leicht bis mittelschwer und führten nicht zum Abbruch der Behandlung. Es kam bei 38,2 % (n = 21) der mit subkutanem Efgartigimod alfa behandelten Patienten zu einer Reaktion an der Injektionsstelle. Wenn es zu Reaktionen an der Injektionsstelle kam, verschwanden diese bei 81,0 % (17/21) der Patienten ohne Behandlung.
In einem gepoolten Datenbestand aus 2 klinischen Studien bei Patienten mit CIDP, die eine kontinuierliche Anwendung von Efgartigimod alfa subkutan erhielten, betrug die Inzidenz von Reaktionen an der Injektionsstelle 26 % (61/235). Eine Analyse in 3-monatigen Intervallen zeigte, dass der Prozentsatz der Teilnehmer mit Reaktionen an der Injektionsstelle in den ersten 3 Monaten der Behandlung am höchsten war (73 Teilnehmer [22,2 %]) und in den folgenden 3-monatigen Intervallen abnahm (Bereich: 0 bis 17 Teilnehmer [6,8 %]).
Infektionen
In der placebokontrollierten Studie ARGX-113-1704 zu gMG mit Efgartigimod alfa intravenös (i.v.) waren die am häufigsten berichteten unerwünschten Wirkungen Infektionen. Insgesamt wurden in dieser 26-wöchigen kontrollierten Phase-III-Studie behandlungsbedingte Infektionen bei 46,4 % (n = 39) der mit Efgartigimod alfa i.v. behandelten Patienten und bei 37,3 % (n = 31) der mit Placebo behandelten Patienten berichtet. Die Zeit vom Behandlungsbeginn bis zum Auftreten von Infektionen betrug 6 Wochen (Medianwert). Die am häufigsten berichteten Infektionen waren Infektionen der oberen Atemwege (bei 10,7 % [n = 9] der mit Efgartigimod alfa intravenös behandelten Patienten und bei 4,8 % [n = 4] der mit Placebo behandelten Patienten) und Harnwegsinfektionen (bei 9,5 % [n = 8] der mit Efgartigimod alfa intravenös behandelten Patienten und bei 4,8 % [n = 4] der mit Placebo behandelten Patienten). In der 10-wöchigen offenen Studie (ARGX-113-2001) mit intravenösem (i.v.) und subkutanem (s.c.) Efgartigimod alfa wurden behandlungsbedingte Infektionen bei 16,4 % (n = 9) der mit Efgartigimod alfa i.v. behandelten Patienten und bei 18,2 % (n = 10) der mit Efgartigimod alfa s.c. behandelten Patienten berichtet. Die am häufigsten berichtete Infektion war eine Harnwegsinfektion (bei 5,5 % [n = 3] der mit Efgartigimod alfa i.v. behandelten Patienten und bei 1,8 % [n = 1] der mit Efgartigimod alfa s.c. behandelten Patienten). Infektionen waren bei Patienten, die Efgartigimod alfa i.v. und s.c. erhielten, meist leicht bis mittelschwer (≤Grad 2 gemäss den Common Terminology Criteria for Adverse Events).
Im placebokontrollierten Teil der Studie ARGX-113-1802 bei Patienten mit CIDP war eine kontinuierliche Anwendung von Efgartigimod alfa subkutan nicht mit einem Anstieg der Inzidenz von Infektionen (31,5 % [35/111] in der Gruppe mit Efgartigimod alfa subkutan und 33,6 % [37/110] in der Placebogruppe) verbunden (siehe Rubrik "Eigenschaften/Wirkungen" ).
Kopfschmerz im Zusammenhang mit dem Verfahren (nur bei intravenöser Anwendung)
Kopfschmerz im Zusammenhang mit dem Verfahren wurde in der placebo kontrollierten 26-wöchigen Phase III Studie bei 4,8 % der mit Efgartigimod alfa intravenös behandelten Patienten und bei 1,2 % der mit Placebo behandelten Patienten berichtet, während dies bei keinem der Patienten in der 10-wöchigen offenen Studie (ARGX-113-2001) mit intravenösem (i.v.) und subkutanem (s.c.) Efgartigimod alfa auftrat. Kopfschmerz im Zusammenhang mit dem Verfahren wurde berichtet, wenn der Kopfschmerz in zeitlichem Zusammenhang mit der intravenösen Infusion von Efgartigimod alfa stand. Alle Ereignisse waren leicht oder mittelschwer, mit Ausnahme eines Ereignisses, das als schwerwiegend (Grad 3) gemeldet wurde.
Anaphylaxie
In verblindeten und offenen klinischen Studien (sowohl mit der intravenösen als auch mit der subkutanen Darreichungsform) wurden keine anaphylaktischen Reaktionen berichtet. Nach der Markteinführung wurden Fälle von anaphylaktischen Reaktionen bei intravenöser Anwendung von Efgartigimod alfa gemeldet. Für Efgartigimod alfa subkutan wurden seit der Markteinführung bislang keine anaphylaktischen Reaktionen berichtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es sind keine spezifischen Anzeichen und Symptome einer Überdosierung mit Efgartigimod alfa bekannt. Im Falle einer Überdosierung sind keine anderen eventuell auftretenden Nebenwirkungen als bei der empfohlenen Dosis zu erwarten. Die Patienten sollten auf Nebenwirkungen überwacht werden und es ist eine geeignete symptomatische und unterstützende Behandlung einzuleiten. Es gibt kein spezifisches Antidot für eine Überdosierung mit Efgartigimod alfa.
Eigenschaften/Wirkungen
ATC-Code
L04AA58
Wirkungsmechanismus
Efgartigimod alfa ist ein Fragment des humanen IgG1-Antikörpers, dessen Affinität für den neonatalen Fc-Rezeptor (FcRn) erhöht wurde. Efgartigimod alfa bindet an FcRn, was zu einer Verringerung der Spiegel von zirkulierendem IgG, einschliesslich pathogener IgG-Autoantikörper, führt. Efgartigimod alfa beeinflusst weder die Spiegel anderer Immunglobuline (IgA, IgD, IgE oder IgM) noch senkt es den Albuminspiegel.
IgG-Autoantikörper sind die zugrunde liegende Ursache der Pathogenese von IgG-vermittelten Autoimmunerkrankungen.
Bei MG beeinträchtigen sie die neuromuskuläre Übertragung, indem sie an Acetylcholin-Rezeptoren (AChR), muskelspezifische Tyrosinkinase (MuSK) oder an das Low-Density-Lipoprotein-Rezeptor-verwandte Protein 4 (LRP4) binden.
Bei CIDP weisen mehrere Evidenzlinien auf die Schlüsselrolle von IgG-Autoantikörpern bei der Pathogenese dieser Krankheit hin. Dazu gehören der Nachweis autoreaktiver IgG-Antikörper gegen Bestandteile myelinisierter Nerven, die passive Übertragung von CIDP-Symptomen auf Tiermodelle mittels Seren oder IgG von CIDP-Patienten sowie die therapeutische Wirkung von Plasmaaustausch und Immunadsorption bei der Behandlung von Patienten mit CIDP.
Pharmakodynamik
Intravenöse Darreichungsform
In der doppelblinden, placebokontrollierten Studie ARGX-113-1704 bei gMG-Patienten verringerte Efgartigimod alfa bei der empfohlenen Dosis von 10 mg/kg und dem empfohlenen Behandlungsplan (einmal wöchentliche Anwendung über 4 Wochen) die IgG-Spiegel und die AChR-Autoantikörperspiegel (AChR-Ab) im Serum (siehe Rubrik "Dosierung/Anwendung" ). Die mittlere prozentuale Abnahme des IgG-Gesamtspiegels im Vergleich zum Ausgangswert erreichte eine Woche nach der letzten Infusion des ersten Behandlungszyklus einen Maximalwert von 61 % und hatte 9 Wochen nach der letzten Infusion wieder den Ausgangswert erreicht. Eine ähnliche Wirkung wurde bei allen IgG-Subtypen beobachtet. Die Abnahme der AChR-Autoantikörperspiegel folgte einem ähnlichen Zeitverlauf mit einer maximalen mittleren prozentualen Abnahme von 58 % eine Woche nach der letzten Infusion und einer Wiederherstellung des Ausgangswerts 7 Wochen nach der letzten Infusion. Ähnliche Veränderungen wurden im zweiten Zyklus der Studie festgestellt.
Subkutane Darreichungsform
In der Studie ARGX-113-2001 folgte die Abnahme der AChR-Ab-Spiegel einem vergleichbaren Zeitverlauf wie der IgG-Gesamtspiegel und war in den mit Efgartigimod alfa subkutan und intravenös behandelten Gruppen ähnlich. In der mit Efgartigimod alfa subkutan behandelten Gruppe und in der mit Efgartigimod alfa intravenös behandelten Gruppe wurde eine Woche nach der letzten Verabreichung eine maximale mittlere prozentuale Abnahme der AChR-Ab-Spiegel von 62,2 % bzw. 59,6 % beobachtet. Sowohl in der mit Efgartigimod alfa subkutan behandelten Gruppe als auch in der mit Efgartigimod alfa intravenös behandelten Gruppe war eine Abnahme des IgG-Gesamtspiegels und des AChR-Ab-Spiegels mit einem klinischen Ansprechen verbunden, gemessen anhand der Veränderung des MG-ADL-Gesamtscores gegenüber Baseline (siehe Abbildung 1).
Abbildung 1. Zusammenhang zwischen Gesamt-IgG und AChR-Ab und MG-ADL-Gesamtscore in der AChR-Ab-seropositiven Population unter Behandlung mit Efgartigimod alfa subkutan (1A) und Efgartigimod alfa intravenös (1B) (Studie ARGX-113-2001)
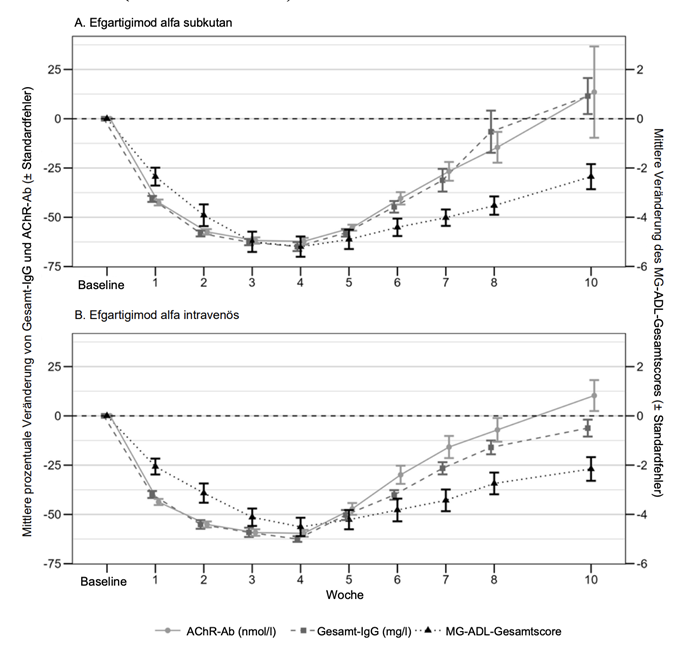
In der Studie ARGX-113-1802 bei Patienten mit CIDP, die eine kontinuierliche, einmal wöchentliche Anwendung von 1000 mg Efgartigimod alfa subkutan erhielten, blieb die mittlere prozentuale Veränderung des Gesamt-IgG-Spiegels gegenüber Baseline ab Woche 4 während des gesamten Behandlungszeitraums erhalten (die mittlere prozentuale Verringerung gegenüber Baseline lag zwischen 66,8 und 71,6 %).
Klinische Wirksamkeit
Generalisierte Myasthenia gravis
Intravenöse Darreichungsform
Die Wirksamkeit von Efgartigimod alfa zur Behandlung von Erwachsenen mit generalisierter Myasthenia gravis (gMG) wurde in einer 26-wöchigen, multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (ARGX-113-1704) untersucht.
In dieser Studie mussten die Patienten beim Screening die folgenden Hauptkriterien erfüllen:
-Klasse II, III oder IV gemäss der Klassifikation der Myasthenia Gravis Foundation of America (MGFA);
-Patienten mit positiven oder negativen serologischen Tests auf Antikörper gegen AChR;
-Gesamtscore von ≥5 im MG-ADL (MG-Activities of Daily Living);
-Behandlung mit stabil eingestellten Dosen einer MG-Therapie vor dem Screening, einschliesslich Acetylcholinesterase (AChE)-Hemmer, Steroiden oder einer nichtsteroidalen immunsuppressiven Therapie (NSIST), entweder in Kombination oder als Monotherapie [NSISTs schlossen unter anderem Azathioprin, Methotrexat, Cyclosporin, Tacrolimus, Mycophenolatmofetil und Cyclophosphamid ein];
-IgG-Werte von mindestens 6 g/l.
Patienten mit gMG der MGFA-Klasse V; Patienten mit dokumentiertem unzureichendem klinischem Ansprechen auf Plasmapherese (plasma exchange, PLEX), Patienten, die einen Monat vor Behandlungsbeginn mit PLEX, intravenösem Immunglobulin (IVIg) und sechs Monate vor Behandlungsbeginn mit monoklonalen Antikörpern behandelt wurden, Patienten, die in den letzten drei Monaten vor Studienbeginn eine Thymektomie erhalten hatten und Patienten mit aktiver (akuter oder chronischer) Hepatitis-B-Infektion, mit Hepatitis-C-Seropositivität, mit AIDS-Diagnose, oder einer schweren Infektion in den letzten 8 Wochen oder einer nicht erfolgreich behandelten malignen Grunderkrankung in den letzten drei Jahren einschliesslich malignem Thymom waren von der Teilnahme an den Studien ausgeschlossen.
Es wurden insgesamt 167 Patienten in die Studie aufgenommen und erhielten nach Randomisierung entweder Efgartigimod alfa intravenös (n = 84) oder Placebo (n = 83). Die Behandlungsgruppen wiesen ähnliche Baseline-Merkmale auf, einschliesslich des medianen Alters bei der Diagnose [45 (19-81) Jahre], des Geschlechts [die meisten waren weiblich; 75 % (Efgartigimod alfa) vs. 66 % (Placebo)], der ethnischen Zugehörigkeit [die meisten Patienten waren weiss; 84,4 %] und des medianen Zeitraums seit der Diagnose [8,2 Jahre (Efgartigimod alfa) und 6,9 Jahre (Placebo)].
Die meisten Patienten (77 % in jeder Gruppe) wurden positiv auf Antikörper gegen AChR (AChR-Ab) getestet, und 23 % der Patienten wurden negativ auf AChR-Ab getestet.
Während der Studie erhielten über 80 % der Patienten in jeder Gruppe AChE-Hemmer, über 70 % in jeder Behandlungsgruppe erhielten Steroide und etwa 60 % in jeder Behandlungsgruppe erhielten eine nichtsteroidale immunsuppressive Therapie (NSIST) in stabiler Dosierung. Zu Beginn der Studie hatten etwa 30 % der Patienten in jeder Behandlungsgruppe vorher noch keine NSIST erhalten.
Der mediane MG-ADL-Gesamtscore betrug in beiden Behandlungsgruppen 9,0, und der mediane Quantitative Myasthenia Gravis(QMG)-Score betrug 17 in der mit Efgartigimod alfa behandelten Gruppe und 16 in der Placebo-Gruppe.
Die Behandlung der Patienten mit Efgartigimod alfa intravenös erfolgte im Rahmen des empfohlenen Dosierungsschemas von 10 mg/kg einmal wöchentlich über 4 Wochen und mit maximal 3 Behandlungszyklen (siehe Rubrik "Dosierung/Anwendung" ).
Die Wirksamkeit von Efgartigimod alfa wurde anhand der Skala MG-ADL (Myasthenia Gravis-Specific Activities of Daily Living) gemessen, mit der die Auswirkungen der gMG auf tägliche Aktivitäten beurteilt wird. Der Gesamtscore liegt im Bereich von 0 bis 24, wobei höhere Werte eine stärkere Beeinträchtigung bedeuten. In dieser Studie war ein MG-ADL-Responder ein Patient mit einer Verringerung des MG-ADL-Gesamtscores um ≥2 Punkte in mindestens 4 aufeinanderfolgenden Wochen im Vergleich zum Wert zu Anfang des Behandlungszyklus, wobei die erste Verringerung nicht später als eine Woche nach der letzten Infusion des Zyklus festzustellen war.
Die Wirksamkeit von Efgartigimod alfa wurde auch anhand des QMG-Scores gemessen, einem System zur Einstufung der Muskelschwäche mit einem möglichen Gesamtscore von 0 bis 39, bei dem höhere Scores eine stärkere Beeinträchtigung bedeuten. In dieser Studie war ein QMG-Responder ein Patient mit einer Verringerung des QMG-Scores um ≥3 Punkte in mindestens 4 aufeinanderfolgenden Wochen im Vergleich zum Wert zu Anfang des Behandlungszyklus, wobei die erste Verringerung nicht später als eine Woche nach der letzten Infusion des Zyklus festzustellen war.
Der primäre Wirksamkeitsendpunkt war der Vergleich des Prozentsatzes der MG-ADL-Responder während des ersten Behandlungszyklus (C1) zwischen den Behandlungsgruppen in der Population der AChR-Ab-seropositiven Patienten.
Ein wichtiger sekundärer Endpunkt war der Vergleich des Prozentsatzes der QMG-Responder während C1 zwischen beiden Behandlungsgruppen bei den AChR-Ab-seropositiven Patienten.
Tabelle 2. MG-ADL- und QMG-Responder in Zyklus 1 in der Population der AChR-Ab-seropositiven Patienten (mITT-Analysegruppe)
Population Efgartigimod alfan/N Placebon/N (%) P-Wert Differenz Efgartigim
(%) od alfa - Placebo
(95 %-KI)
MG-ADL AChR-Ab-seropositiv 44/65 (67,7) 19/64 (29,7) <0,0001 38,0 (22,1; 54,0)
QMG AChR-Ab-seropositiv 41/65 (63,1) 9/64 (14,1) <0,0001 49,0 (34,5; 63,5)
AChR-Ab = Anti-Acetylcholin-Rezeptor-Antikörper; MG-ADL = Myasthenia Gravis Activities of Daily Living (Fragebogen zur Beurteilung der MG-Krankheitsaktivität); QMG = Quantitative Myasthenia Gravis (Fragebogen zur Bestimmung eines Quantitativen Myasthenia Gravis-Score); mITT = modifizierte Intent-to-treat-Gruppe; n = Anzahl der Patienten, bei denen die jeweilige Beobachtung gemacht wurde; N = Anzahl der Patienten in der Analysegruppe; KI = Konfidenzintervall;
Logistische Regression, stratifiziert nach AChR-Ab-Status (sofern zutreffend), Japanisch/Nicht-Japanisch und Standardbehandlung, mit MG-ADL-/QMG-Baseline-Score als Kovariate
Zweiseitiger exakter p-Wert
Analysen zeigen, dass die MG-ADL-Responderraten während des zweiten Behandlungszyklus ähnlich waren wie im ersten Behandlungszyklus (siehe Tabelle 3).
Tabelle 3. MG-ADL- und QMG-Responder in Zyklus 2 in der Population der AChR-Ab-seropositiven Patienten (mITT-Analysegruppe)
Population Efgartigimod alfa n/N (%) Placebo n/N (%) MG-ADL AChR-Ab-seropositiv 36/51 (70,6) 11/43 (25,6) QMG AChR-Ab-seropositiv 24/51 (47,1) 5/43 (11,6)
AChR-Ab = Anti-Acetylcholin Rezeptor-Antikörper; MG-ADL = Myasthenia Gravis Activities of Daily Living (Fragebogen zur Beurteilung der MG-Krankheitsaktivität); QMG = Quantitative Myasthenia Gravis (Fragebogen zur Bestimmung eines Quantitativen Myasthenia Gravis-Score); mITT = modifizierte Intent-to-treat-Gruppe; n = Anzahl der Patienten, bei denen die jeweilige Beobachtung gemacht wurde; N = Anzahl der Patienten in der Analysegruppe.
Bei Patienten mit einer Thymektomie in der Vorgeschichte waren 27 (60 %) Patienten in der Efgartigimod alfa-Gruppe MG-ADL-Responder im Vergleich zu 8 (27 %) Patienten in der Placebo-Gruppe.
Exploratorische Daten zeigen, dass bei 37/44 (84 %) der mit Efgartigimod alfa intravenös behandelten Patienten der AChR-Ab-seropositiven MG-ADL-Responder ein Ansprechen innerhalb von 2 Wochen nach der ersten Infusion beobachtet wurde.
In der doppelblinden, placebokontrollierten Studie (ARGX-113-1704) konnte gemäss klinischem Studienprotokoll ein nachfolgender Behandlungszyklus erst eingeleitet werden, wenn alle folgenden Kriterien erfüllt waren:
(1) die Mindestzeit zwischen den Behandlungszyklen betrug 8 Wochen ab der ersten Infusion des vorherigen Zyklus;
(2) der Patient wies einen MG-ADL-Gesamtscore von ≥5 Punkten mit >50 % des Gesamtscores durch nicht-okuläre Symptome auf und
(3) nur für Patienten, die im vorangegangenen Behandlungszyklus den Responder Status (Definition s. oben) erreicht haben und nun einen Verlust des Ansprechens (definiert als eine Verringerung des MG-ADL-Gesamtscores <2 Punkte im Vergleich zum entsprechenden Zyklus-Ausgangs-Wert) zeigen.
In der Gesamtpopulation betrug die mittlere Zeit bis zum zweiten Behandlungszyklus in der Efgartigimod alfa intravenös behandelten Gruppe 13 Wochen (Standardabweichung 5,5 Wochen) und die mediane Zeit 10 Wochen (8-26 Wochen) ab der ersten Infusion im ersten Behandlungszyklus.
Bei Patienten, die auf die Behandlung ansprachen (Verringerung des MG-ADL-Gesamtscores um ≥2 Punkte vs Zyklus-Baseline), betrug die Dauer der klinischen Besserung bei 5/44 Patienten (11 %) 5 Wochen, bei 14/44 Patienten (32 %) 6-7 Wochen, bei 10/44 Patienten (23 %) 8-11 Wochen und bei 15/44 Patienten (34 %) 12 Wochen oder mehr.
Subkutane Darreichungsform
Es wurde eine 10-wöchige, randomisierte, offene, multizentrische Parallelgruppenstudie (ARGX-113-2001) bei erwachsenen Patienten mit gMG durchgeführt, um die Nichtunterlegenheit der pharmakodynamischen Wirkung von Efgartigimod alfa subkutan im Vergleich zu Efgartigimod alfa intravenös zu bewerten. Die wichtigsten Ein- und Ausschlusskriterien waren dieselben wie in der Studie ARGX-113-1704.
Es wurden insgesamt 110 Patienten randomisiert und erhielten über 4 Wochen einen Zyklus mit einmal wöchentlicher Verabreichung von entweder 1000 mg Efgartigimod alfa subkutan (n = 55) oder 10 mg/kg Efgartigimod alfa intravenös (n = 55). Die meisten Patienten waren positiv auf Antikörper gegen AChR (AChR-Ab): 45 Patienten (82 %) in der mit Efgartigimod alfa subkutan behandelten Gruppe und 46 Patienten (84 %) in der mit Efgartigimod alfa intravenös behandelten Gruppe. Alle Patienten erhielten vor dem Screening stabile Dosen einer MG-Therapie, die AChE-Hemmer, Steroide oder NSIST, entweder in Kombination oder als Monotherapie, umfasste.
Die Baseline-Merkmale in den Behandlungsgruppen waren vergleichbar.
Während der Studie erhielten über 80 % der Patienten in jeder Gruppe AChE-Hemmer, über 60 % der Patienten in jeder Gruppe erhielten Steroide und etwa 40 % in jeder Behandlungsgruppe erhielten NSIST in stabilen Dosen. Bei der Aufnahme in die Studie hatten etwa 56 % der Patienten in jeder Behandlungsgruppe zuvor noch keine NSIST erhalten.
Der primäre Endpunkt war der Vergleich der prozentualen Verringerung der IgG-Gesamtspiegel gegenüber dem Baseline-Wert an Tag 29 zwischen den Behandlungsgruppen in der Gesamtpopulation. Die Ergebnisse in der AChR-Ab-seropositiven Population zeigen, dass Efgartigimod alfa subkutan gegenüber Efgartigimod alfa intravenös nicht unterlegen ist (siehe Tabelle 4).
Tabelle 4. ANCOVA der prozentualen Veränderung des IgG-Gesamtspiegels gegenüber Baseline an Tag 29 in der AChR-Ab-seropositiven Population (mITT-Analysegruppe)
Efgartigimod alfa Efgartigimod alfa Differenz Efgartigim
s.c. i.v. od alfa s.c. -
Efgartigimod alfa
i.v.
N LS-Mittelwert 95 %-KI N LS-Mittelwert 95 %-KI Differenz der 95 %-KI p-Wert
LS-Mittelwerte
41 -66,9 -69,78; -64,02 43 -62,4 -65,22; -59,59 -4,5 -8,53; -0,46 <0,0001
AChR-Ab = Anti-Acetylcholin-Rezeptor-Antikörper; ANCOVA = Kovarianzanalyse; KI = Konfidenzintervall; s.c. = subkutan; i.v. = intravenös; LS = Least Squares (Kleinstquadrate); mITT = modifizierte Intent-to-treat-Analysegruppe; N = Anzahl der mittels ANCOVA analysierten Patienten
Sekundäre Wirksamkeitsendpunkte waren Vergleiche des Prozentsatzes der MG-ADL- und QMG-Responder, wie in der Studie ARGX-113-1704 definiert, zwischen beiden Behandlungsgruppen. Die Ergebnisse in der AChR-Ab-seropositiven Population sind in Tabelle 5 dargestellt.
Tabelle 5. MG-ADL- und QMG-Responder an Tag 29 in der AChR-Ab-seropositiven Population (mITT-Analysegruppe)
Efgartigimod alfa Efgartigimod alfa Differenz Efgartigimod alfa
s.c.n/N (%) i.v.n/N (%) s.c.-Efgartigimod alfa i.v. (95
%-KI)
MG-ADL 32/45 (71,1) 33/46 (71,7) -0,6 (-19,2 bis 17,9)
QMG 31/45 (68,9) 24/45 (53,3) 15,6 (-4,3 bis 35,4)
AChR-Ab = Anti-Acetylcholin-Rezeptor-Antikörper; MG-ADL = Myasthenia Gravis Activities of Daily Living (Fragebogen zur Beurteilung der MG-Krankheitsaktivität); QMG = Quantitative Myasthenia Gravis (Fragebogen zur Bestimmung eines Quantitativen Myasthenia Gravis-Scores); s.c. = subkutan; i.v. = intravenös; mITT = modifizierte Intent-to-treat-Analysegruppe; n = Anzahl der Patienten, bei denen die jeweilige Beobachtung gemacht wurde; N = Anzahl der Patienten in der Analysegruppe; KI = Konfidenzintervall;
Exploratorische Daten zeigen, dass unter den AChR-Ab-seropositiven MG-ADL-Respondern bei 28/32 Patienten (88 %), die mit Efgartigimod alfa subkutan behandelt wurden, und bei 27/33 Patienten (82 %), die mit Efgartigimod alfa intravenös behandelt wurden, innerhalb von 2 Wochen nach der ersten Verabreichung ein Ansprechen erfolgte.
Chronisch-entzündliche demyelinisierende Polyneuropathie
Die Wirksamkeit von Efgartigimod alfa subkutan zur Behandlung von Erwachsenen mit CIDP wurde in der prospektiven, multizentrischen Studie ARGX-113-1802 untersucht, die in 2 Behandlungsphasen durchgeführt wurde: eine offene Phase A und eine doppelblinde, placebokontrollierte Phase B mit randomisiertem Absetzen.
Die Patienten hatten in den letzten 6 Monaten vor Studienaufnahme entweder eine CIDP-Behandlung erhalten oder nicht. Diejenigen, die zuvor eine CIDP-Behandlung erhalten hatten, sowie diejenigen, die keine CIDP-Behandlung erhalten hatten und bei denen keine Hinweise auf eine kürzlich eingetretene Verschlechterung der CIDP vorlagen, traten in eine behandlungsfreie Vorlaufphase ein. Patienten, bei denen Hinweise auf eine klinisch bedeutsame Verschlechterung vorlagen, nahmen anschliessend an Phase A der Studie teil. Diejenigen Patienten ohne CIDP-Behandlung, bei denen vor kurzem eine Verschlechterung der CIDP nachgewiesen wurde, liessen die Vorlaufphase aus und traten direkt in Phase A ein.
Insgesamt wurden 322 Patienten in Phase A aufgenommen. Die Patienten erhielten bis zu 12 einmal wöchentliche Injektionen von Efgartigimod alfa subkutan in einer Dosierung von 1000 mg, bis bei 2 aufeinanderfolgenden Studienbesuchsterminen eine bestätigte klinische Verbesserung (ECI, evidence of clinical improvement) festgestellt wurde. Anschliessend traten die Patienten mit bestätigter ECI in Phase B der Studie ein und erhielten nach dem Zufallsprinzip entweder wöchentlich Efgartigimod alfa subkutan (111 Patienten) oder Placebo (110 Patienten). Eine ECI war definiert als klinische Verbesserung auf der Skala Adjusted Inflammatory Neuropathy Cause and Treatment (aINCAT) oder als Verbesserung auf der Inflammatory Rasch-built Overall Disability Scale (I-RODS)/Grip Strength bei Patienten, die sich vor Phase A nur auf diesen Skalen verschlechtert hatten.
In Phase A hatten die Patienten ein medianes Alter von 54 Jahren (Bereich: 20 bis 82 Jahre), einen medianen Zeitraum seit der CIDP-Diagnose von 2,8 Jahren und einen medianen INCAT-Score von 4,0. 65 Prozent waren männlich und 66 % waren weiss. In Phase B hatten die Patienten ein medianes Alter von 55 Jahren (Bereich: 20 bis 82 Jahre), einen medianen Zeitraum seit der CIDP-Diagnose von 2,2 Jahren und einen medianen INCAT-Score von 3,0. 64 Prozent waren männlich und 65 % waren weiss. Die Merkmale bei Baseline in Phase B waren zwischen den Behandlungsgruppen ähnlich.
In Phase A war der primäre Endpunkt der Prozentsatz der Responder, d.h. der Patienten, die eine bestätigte ECI erreichten. Der primäre Endpunkt wurde bei 66,5 % der Patienten erreicht; weitere Einzelheiten sind in Tabelle 6 aufgeführt.
Ein sekundärer Endpunkt in Phase A war die Zeit bis zur ersten bestätigten ECI. Woche 4 war der früheste Zeitpunkt, zu dem die Kriterien für eine ECI erfüllt werden konnten. Zu diesem Zeitpunkt erreichten bis zu 40 % der Patienten eine ECI. Auf der Grundlage einer zusätzlichen, vorab festgelegten Analyse zeigten 25 % der Patienten nach 9 Tagen eine klinisch relevante Verbesserung bei mindestens einem der 3 Parameter (aINCAT, I-RODS oder Grip Strength).
Die Mehrzahl der Patienten erreichte eine bestätigte ECI in allen vorherigen CIDP-Medikationsgruppen.
Tabelle 6. Nachweis der klinischen Verbesserung bei Patienten mit CIDP in Phase A der Studie ARGX-113-1802
ECI-Responder und Zeit bis zur ersten bestätigten ECI Phase A Efgartigimod alfa s.c. (N = 322) ECI-Responder (Patienten mit bestätigter klinischer Verbesserung) n/N 214/322 (66,5 %) (61,0; (%) (95 %-KI) 71,6) Zeit bis zur ersten bestätigten ECI in Tagen Median (95 %-KI) 43,0 (31,0; 51,0)
n = Anzahl der Patienten, für die die Beobachtung gemeldet wurde; N = Anzahl der Patienten in der Analysegruppe
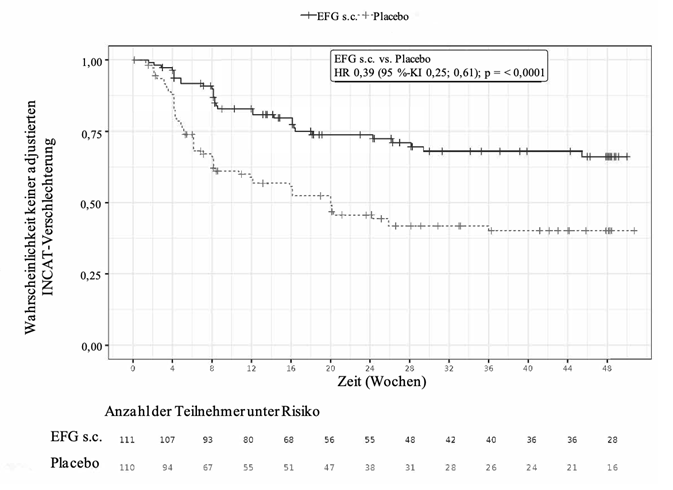
In Phase B war der primäre Endpunkt definiert als die Zeit bis zum Auftreten des ersten Nachweises einer klinischen Verschlechterung (ein Anstieg des aINCAT-Wertes um 1 Punkt im Vergleich zu Baseline von Phase B, der bei einem nachfolgenden Besuchstermin nach dem ersten Anstieg des aINCAT-Wertes um 1 Punkt bestätigt wurde, oder ein Anstieg des aINCAT-Wertes um ≥2 Punkte im Vergleich zu Baseline von Phase B). Patienten, die Efgartigimod alfa subkutan erhielten, blieben im Vergleich zu Patienten, die Placebo erhielten, signifikant länger rezidivfrei (d.h. ohne klinische Verschlechterung), was durch eine Hazard Ratio von 0,394 [95 %-KI (0,253; 0,614)] belegt wurde. Bei 31/111 Patienten (27,9 %), die in Phase B der Studie Efgartigimod alfa subkutan erhielten, kam es zu einem Rezidiv, verglichen mit 59/110 Patienten (53,6 %), die Placebo erhielten. Die Ergebnisse sind in Tabelle 7 und Abbildung 2 dargestellt.
Tabelle 7. Erster Nachweis der klinischen Verschlechterung bei Patienten mit CIDP in Phase B der Studie ARGX-113-1802
Zeit bis zum 1. aINCAT-Anstieg (klinische Phase B
Verschlechterung)
Efgartigimod alfa s.c. (N = 111) Placebo (N = 110)
Hazard Ratio (95 %-KI) 0,394 (0,253; 0,614) p-Wert
<0,0001
Mediane Zeit in Tagen (95 %-KI) NC (NC; NC) 140,0 (75,0; NC)
NC = nicht berechnet; N = Anzahl der Patienten in der Analysegruppe; aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment
Abbildung 2. Zeit bis zur ersten aINCAT-Verschlechterung (Kaplan-Meier-Kurve) bei Patienten mit CIDP in Phase B der Studie ARGX-113-1802
Pharmakokinetik
Absorption
Basierend auf der Populations-PK-Datenanalyse beträgt die geschätzte Bioverfügbarkeit bei Anwendung von Efgartigimod alfa 1000 mg subkutan 77 %.
Der mittlere Ctrough-Wert nach 4 einmal wöchentlichen Anwendungen von Efgartigimod alfa 1000 mg subkutan und Efgartigimod alfa 10 mg/kg intravenös betrug 22,0 µg/ml (37 % VK) bzw. 14,9 µg/ml (43 % VK). Die AUC0-168Std. von Efgartigimod alfa nach Anwendung eines Behandlungszyklus mit 1000 mg subkutan und 10 mg/kg intravenös waren vergleichbar.
Bei Patienten, die eine kontinuierliche subkutane Anwendung von 1000 mg Efgartigimod alfa einmal wöchentlich erhielten, lag der mittlere Ctrough-Wert zwischen 14,9 und 20,1 µg/ml.
Distribution
Basierend auf der Populations-PK-Datenanalyse bei gesunden Probanden und Patienten beträgt das Verteilungsvolumen 18 l.
Metabolismus
Efgartigimod alfa wird voraussichtlich durch proteolytische Enzyme in kleine Peptide und Aminosäuren abgebaut.
Elimination
Die terminale Halbwertszeit beträgt 80 bis 120 Stunden (3 bis 5 Tage). Basierend auf der Populations-PK-Datenanalyse beträgt die Clearance 0,128 l/h. Die Molekülmasse von Efgartigimod alfa beträgt ungefähr 54 kDa, was im Grenzbereich der renalen Filtration von Molekülen liegt.
Linearität/Nicht-Linearität
Das pharmakokinetische Profil von Efgartigimod alfa ist linear, dosis- oder zeitunabhängig, mit minimaler Akkumulation.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurden keine spezielle pharmakokinetische Studie bei Patienten mit Leberfunktionsstörung durchgeführt.
Die Untersuchung des Einflusses von Leberfunktionsmarkern als Kovariate in einer populationspharmakokinetischen Analyse ergab keine Auswirkungen auf die Pharmakokinetik von Efgartigimod alfa.
Nierenfunktionsstörungen
Es wurden keine speziellen pharmakokinetischen Studien bei Patienten mit Nierenfunktionsstörung durchgeführt.
Die Untersuchung des Einflusses der mithilfe von Nierenfunktionsmarkern geschätzten glomerulären Filtrationsrate [eGFR] als Kovariate in einer populationspharmakokinetischen Analyse ergab bei Patienten mit leichter Nierenfunktionsstörung (eGFR 60-89 ml/min/1,73 m2) einen Anstieg der Exposition (11 % zu 21 %) . Bei Patienten mit leichter Nierenfunktionsstörung wird keine spezifische Dosisanpassung empfohlen.
Es liegen keine ausreichenden Daten zu den Auswirkungen einer mässig eingeschränkten Nierenfunktion (eGFR 30-59 ml/min/1,73 m2) und einer schweren Nierenfunktionsstörung (eGFR <30 ml/min/1,73 m2) auf die pharmakokinetischen Parameter von Efgartigimod alfa vor.
Alter, Geschlecht, Ethnie und Körpergewicht
Die Pharmakokinetik von Efgartigimod alfa wurde durch Alter (19-84 Jahre), Geschlecht, Ethnie und Körpergewicht nicht beeinflusst.
Präklinische Daten
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie und Toxizität bei wiederholter Gabe lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
In Reproduktionsstudien an Ratten und Kaninchen führte die intravenöse Verabreichung von Efgartigimod alfa in Dosierungen bis zum 11-Fachen (Ratten) bzw. 56-Fachen (Kaninchen) der Exposition (AUC) bei der maximal empfohlenen therapeutischen Dosis (10 mg/kg) weder zu unerwünschten Wirkungen auf die Fertilität und Trächtigkeit noch wurden teratogene Wirkungen festgestellt.
Karzinogenität und Genotoxizität
Es wurden keine Studien zur Beurteilung des karzinogenen und genotoxischen Potenzials von Efgartigimod alfa durchgeführt.
Hyaluronidase kommt in den meisten Geweben des menschlichen Körpers vor. Basierend auf den konventionellen Studien zur Toxizität bei wiederholter Gabe, einschliesslich sicherheitsbezogener pharmakologischer Endpunkte, lassen die präklinischen Daten für rekombinante humane Hyaluronidase keine besonderen Gefahren für den Menschen erkennen. Reproduktionstoxikologische Studien mit rekombinanter humaner Hyaluronidase zeigten bei Mäusen bei hoher systemischer Exposition embryofetale Toxizität, jedoch kein teratogenes Potenzial.
Sonstige Hinweise
Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf Vyvgart, Injektionslösung / Vyvgart, Injektionslösung in einer Fertigspritze nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Vyvgart, Injektionslösung
Ungeöffnete Durchstechflaschen können gegebenenfalls bis zu 3 Tage bei Raumtemperatur (bis zu 30 °C) gelagert werden. Nach der Lagerung bei Raumtemperatur können ungeöffnete Durchstechflaschen wieder in den Kühlschrank gestellt werden. Wenn sie ausserhalb des Kühlschranks gelagert und dann wieder in den Kühlschrank gestellt werden, sollte die Gesamtdauer der Lagerung ausserhalb des Kühlschranks 3 Tage nicht überschreiten.
Aus mikrobiologischen Gründen sollte die gebrauchsfertige Zubereitung sofort verwendet werden, es sei denn die Methode der Vorbereitung der Spritze schliesst das Risiko einer mikrobiellen Kontamination aus. Wenn die gebrauchsfertige Zubereitung nicht sofort verwendet wird, ist der Anwender für die Dauer und die Bedingungen der Aufbewahrung verantwortlich.
Vyvgart, Injektionslösung in einer Fertigspritze
Patienten können die ungeöffnete Fertigspritze bei Raumtemperatur in der Originalverpackung bei bis zu 30 °C für einen Zeitraum von bis zu 1 Monat nach Entnahme aus dem Kühlschrank oder bis zum Verfallsdatum aufbewahren, je nachdem, was zuerst eintritt.
Aus mikrobiologischen Gründen sollte die gebrauchsfertige Zubereitung sofort verwendet werden. Wenn die gebrauchsfertige Zubereitung nicht sofort verwendet wird, ist der Anwender für die Dauer und die Bedingungen der Aufbewahrung verantwortlich.
Besondere Lagerungshinweise
Im Kühlschrank lagern (2 - 8 °C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Nicht schütteln.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung – Vyvgart, Injektionslösung
Vyvgart liegt als gebrauchsfertige Lösung in einer Durchstechflasche zur einmaligen Anwendung vor. Das Arzneimittel muss nicht verdünnt werden.
Per Sichtprüfung kontrollieren, ob der Inhalt der Durchstechflasche eine gelbliche, klare bis opaleszierende Lösung und frei von Partikeln ist. Wenn sichtbare Partikel vorhanden sind, darf die Durchstechflasche nicht verwendet werden.
Nach der Entnahme der Durchstechflasche aus dem Kühlschrank mindestens 15 Minuten warten, bevor die Injektion durchgeführt wird, damit die Lösung Raumtemperatur annehmen kann (siehe Abschnitt "Haltbarkeit" ).
Die Injektionslösung kann unter Verwendung einer Polypropylenspritze, Transferkanülen aus Edelstahl und eines geflügelten Infusionssets aus Polyvinylchlorid mit einem maximalen Füllvolumen von 0,4 ml verabreicht werden.
-Den gesamten Inhalt der Efgartigimod alfa-Lösung mit einer Transferkanüle aus der Durchstechflasche aufziehen.
-Die Kanüle an der Spritze gegen das geflügelte Infusionsset austauschen.
-Vor der Injektion sollte das Volumen in der Spritze auf 5,6 ml gebracht werden.
Hinweise für die Handhabung – Vyvgart, Injektionslösung in einer Fertigspritze
Vyvgart liegt als gebrauchsfertige Lösung in einer Fertigspritze zur einmaligen Anwendung vor. Das Arzneimittel muss nicht verdünnt werden.
Per Sichtprüfung kontrollieren, ob der Inhalt der Fertigspritze eine gelbliche, klare bis opaleszierende Färbung hat und frei von Partikeln ist. Wenn sichtbare Partikel vorhanden sind, darf die Fertigspritze nicht verwendet werden.
Nach der Entnahme der Fertigspritze aus dem Kühlschrank mindestens 30 Minuten warten, bevor die Injektion durchgeführt wird, damit die Lösung Raumtemperatur annehmen kann (siehe Rubrik "Dosierung/Anwendung" ). Nach der Vorbereitung zur Injektion sollte sie sofort angewendet werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Anforderungen zu beseitigen.
Zulassungsnummer
69725, 70518 (Swissmedic)
Packungen
Vyvgart, Injektionslösung
5,6 ml Lösung in einer 6ml- Durchstechflasche aus Typ-I-Glas mit Gummistopfen, Aluminiumversiegelung und Flip-off-Schutzkappe aus Polypropylen.
Packungen mit einer Durchstechflasche. (A)
Vyvgart, Injektionslösung in einer Fertigspritze
5 ml Lösung in einer Fertigspritze (Typ-I-Glas) zur einmaligen Anwendung mit Gummistopfen und Gummikappe für die Spitze.
Packungsgrössen:
1 Fertigspritze. (A)
4 Fertigspritzen. (A)
Zulassungsinhaberin
argenx Switzerland SA, 1214 Vernier
Stand der Information
Januar 2026