Eigenschaften/WirkungenATC-Code
L04AA34
Pharmakotherapeutische Gruppe: Selektive Immunsuppressiva.
Wirkungsmechanismus
Lemtrada ist ein rekombinanter, aus DNS abgeleiteter, humanisierter monoklonaler Antikörper, der sich gegen das 21- bis 28-kD-Glykoprotein CD52 auf der Zelloberfläche richtet. Lemtrada ist ein IgG1-Kappa-Antikörper mit humanem variablem Gerüst und humanen konstanten Regionen und komplementär-determinierenden Regionen eines murinen (Ratte) monoklonalen Antikörpers. Der Antikörper hat ein ungefähres Molekulargewicht von 150 kD.
Lemtrada bindet an CD52, ein Oberflächenantigen, das auf T-(CD3+-) und B-(CD19+-) Lymphozyten in hohen Konzentrationen und auf natürlichen Killerzellen, Monozyten und Makrophagen in niedrigeren Konzentrationen vorhanden ist. Auf neutrophilen Granulozyten, Plasmazellen und Knochenmarkstammzellen ist CD52 in geringen Konzentrationen oder nicht nachweisbar. Lemtrada übt seine Wirkung nach Bindung an die Oberfläche von T- und B-Lymphozyten über eine antikörperabhängige Lyse der Lymphozyten und eine komplementvermittelte Lyse aus.
Der Mechanismus der therapeutischen Wirkung von Lemtrada bei der MS ist noch nicht vollständig aufgeklärt. Allerdings weisen wissenschaftliche Untersuchungen auf immunmodulierende Wirkungen im Zusammenhang mit einer initialen Depletion und anschliessenden Repopulation von Lymphozyten hin, wie zum Beispiel:
·Änderungen von Anzahl, prozentualem Anteil und Eigenschaften einiger Lymphozyten-Subpopulationen nach der Behandlung
·Vermehrtes Auftreten von regulatorischen T-Zell-Populationen
·Vermehrtes Auftreten von T- und B-Gedächtniszellen
·Vorübergehende Wirkungen auf bestimmte Zellen des angeborenen Immunsystems (d.h. neutrophile Granulozyten, Makrophagen, NK-Zellen)
Die durch Lemtrada bewirkte Depletion der zirkulierenden B- und T-Zellen und die nachfolgende Repopulation können das Schubpotenzial verringern und damit schliesslich das Fortschreiten der Erkrankung hinauszögern.
Pharmakodynamische Wirkungen
Lemtrada bewirkt im Anschluss an jeden Behandlungszyklus eine Depletion von zirkulierenden T- und B-Lymphozyten, wobei der niedrigste Wert etwa 1 Monat nach dem Behandlungszyklus gemessen wird (d.h. bei der ersten Untersuchung nach der Behandlung in den klinischen Phase-3-Studien). Mit der Zeit kommt es zu einer Repopulation der Lymphozyten und die B-Zell-Zahlen sind normalerweise innerhalb von 6 Monaten wieder hergestellt. Die Zahlen der T-Lymphozyten kehren langsamer auf den Normwert zurück und in der Regel sind die Ausgangswerte 12 Monate nach der Behandlung noch nicht wieder erreicht. Sechs Monate nach einem Behandlungszyklus wiesen etwa 40% der Patienten und 12 Monate nach einem Behandlungszyklus etwa 80% der Patienten Gesamtlymphozytenzahlen im Bereich der Untergrenze des Normbereichs auf.
Lemtrada hat nur vorübergehenden Einfluss auf neutrophile Granulozyten, Monozyten, eosinophile Granulozyten, basophile Granulozyten und natürliche Killerzellen.
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Lemtrada bei Patienten mit RRMS wurde in 3 randomisierten, Bewerter-verblindeten klinischen Studien mit aktiver Vergleichsgruppe sowie in einer nicht-kontrollierten, Bewerter-verblindeten Verlängerungsstudie bei MS-Patienten beurteilt.
Die Studien 1 und 2 (CAMMS323/CARE-MS I bei nicht vorbehandelten Patienten und CAMMS32400507/CARE-MS II bei Patienten mit unzureichendem Ansprechen auf die vorherige Behandlung) schlossen Patienten mit aktiver schubförmig-remittierender MS ein, die in den vorangegangenen 2 Jahren mindestens 2 klinische Schubereignisse hatten. Neurologische Untersuchungen erfolgten alle 12 Wochen sowie bei Verdacht auf einen Schub. Einmal jährlich erfolgte eine Beurteilung mittels MRT. Die Beobachtungsdauer betrug 2 Jahre. In beiden Studien wurden die Patienten randomisiert einer Behandlung mit Lemtrada 12 mg/Tag als einmal tägliche i.v. Infusion über 5 Tage in Monat 0 und 3 Tage in Monat 12 (die 12-mg-Gruppe) oder IFNB-1a 44 µg als dreimal wöchentliche s.c. Injektion zugeteilt. Studie 2 umfasste darüber hinaus einen explorativen Behandlungsarm mit Lemtrada 24 mg/Tag als einmal tägliche i.v. Infusion über 5 Tage in Monat 0 und 3 Tage in Monat 12 (die 24-mg-Gruppe). Primäre Endpunkte der Studien 1 und 2 waren die über 2 Jahre berechnete jährliche Schubrate (ARR) und die Zeit bis zur bestätigten Verschlechterung der Behinderung (CDW für engl. «Confirmed Disability Worsening»), welche definiert war als über 6 Monate anhaltender Anstieg des Punktwerts auf der Expanded Disability Status Scale (EDSS) von einem Ausgangswert von ≥1,0 um mindestens 1 Punkt (bzw. bei Patienten mit einem EDSS-Ausgangswert von 0 um 1,5 Punkte).
Studie 1 (CAMMS323) schloss Patienten mit aktiver RRMS (mindestens 2 Schübe in den vorangegangenen 2 Jahren) und einem EDSS von 0–3,0 ein (n = 376 für Lemtrada 12 mg und n = 187 für IFNB-1a). Bei Studienbeginn betrug das Durchschnittsalter 33 Jahre, die mittlere Erkrankungsdauer 2 Jahre und der mittlere EDSS-Score 2,0. Die Patienten hatten vor ihrem Einschluss in die Studie noch keine Therapie gegen ihre MS erhalten.
Nach 2 Jahren war die ARR bei den mit Lemtrada behandelten Patienten im Vergleich zu den mit IFNB-1a behandelten Patienten signifikant um 55% verringert. Die über 6 Monate anhaltende bestätigte Verschlechterung der Behinderung (CDW) unterschied sich nicht statistisch signifikant zwischen den beiden Behandlungsgruppen: 8% der mit Lemtrada und 11% der mit IFNB-1a behandelten Patienten wiesen einen anhaltenden Anstieg des EDSS-Scores auf. Der Einfluss der Behandlung auf klinische Endpunkte wurde durch einen signifikanten Effekt auf MRT-Parameter der Entzündung und Krankheitsprogression unterstützt, wie unter anderem das Gehirnvolumen.
Die Ergebnisse sind in Tabelle 2 dargestellt.
Tabelle 2: Wichtige klinische und MRT-Endpunkte aus Studie 1
|
Endpunkt
|
Lemtrada
(n=376)
|
IFNB-1a s.c.
(n=187)
| |
Klinische Endpunkte
| |
Schubrate (koprimärer Endpunkt)1
| |
Patienten mit Ereignis (Anzahl der Ereignisse)
|
82 (119)
|
75 (122)
| |
ARR (95%-KI)
|
0,18
(0,13; 0,23)
|
0,39
(0,29; 0,53)
| |
Relatives Risiko (95%-KI)
|
0,45
(0,32; 0,63)
|
| |
Risikoreduktion
|
54,88
|
| |
p-Wert
|
<0,0001
|
| |
Behinderung (CDW ≥6 Monate; koprimärer Endpunkt)2
| |
Schätzung der Patienten mit einer 6-monatigen CDW (95%-KI)
|
8,00
(5,66; 11,24)
|
11,12
(7,32; 16,71)
| |
Relatives Risiko (95%-KI)
|
0,70
(0,40; 1,23)
|
| |
p-Wert
|
0,2173
|
| |
Anteil der nach 2 Jahren schubfreien Patienten (%)
| |
Schätzung (95%-KI)
|
77,59
(72,87; 81,60)
|
58,69
(51,12; 65,50)
| |
p-Wert
|
<0,0001
|
| |
Änderung des EDSS gegenüber dem Ausgangswert nach 2 Jahren
| |
Schätzung (95%-KI)
|
-0,14
(-0,25; -0,02)
|
-0,14
(-0,29; 0,01)
| |
p-Wert
|
0,4188
|
| |
MRT-Endpunkte
| |
Änderung des T2-Läsionsvolumens gegenüber dem Ausgangswert nach 2 Jahren (%)
|
-9,3
(-19,6; -0,2)
|
-6,5
(-20,7; 2,5)
| |
p-Wert
|
0,3080
|
| |
Patienten mit neuen / sich vergrössernden T2-Läsionen bis Jahr 2 (%)
|
48,5 (23,1)
|
57,6 (40,6)
| |
p-Wert
|
0,0352
|
| |
Patienten mit Gadolinium-anreichernden Läsionen bis Jahr 2 (%)
|
15,4
|
27,0
| |
p-Wert
|
0,0008
|
| |
Patienten mit neuen hypointensen T1-Läsionen bis Jahr 2 (%)
|
24,0
|
31,4
| |
p-Wert
|
0,0545
|
| |
Änderung der Hirnatrophie gegenüber dem Ausgangswert nach 2 Jahren (%)
|
-0,867
|
-1,488
| |
p-Wert
|
<0,0001
|
|
Beim EDSS-Score ist die mittlere Änderung angegeben, ermittelt mit einem gemischten Modell für wiederholte Messungen. Beim T2-Läsionsvolumen in der MRT und bei der Gehirn-Parenchym-Fraktion sind die medianen Änderungen angegeben.
1 Koprimärer Endpunkt: ARR und CDW. Die Studie wurde als erfolgreich bewertet, wenn wenigstens einer der beiden koprimären Endpunkte erreicht wurde.
2 Die Zeit bis zum Auftreten von CDW war definiert als Anstieg um mindestens 1 Punkt auf der erweiterten Kurtzke-Skala (Expanded Disability Status Scale, EDSS), ausgehend von einem Baseline-EDSS-Wert von ≥1,0 (Anstieg um 1,5 Punkte bei Patienten mit einem Baseline-EDSS-Wert von 0), über 6 Monate anhaltend.
Ergänzende Analysen zeigten, dass Lemtrada 12 mg/Tag im Einklang mit seiner Wirkung auf die Schubrate auch zu einer signifikanten Reduktion der Patienten mit schwerem Schub (Reduktion um 61%; p = 0,0056) und der mit Kortikosteroiden behandelten Schübe (Reduktion um 58%, p < 0,0001) im Vergleich zu IFNB-1a führte.
Studie 2 (CAMMS32400507) schloss Patienten mit aktiver RRMS (mindestens 2 Schübe in den vorangegangenen 2 Jahren) und einem EDSS von 0–5 ein (n = 426 für Lemtrada 12 mg und n = 202 für IFNB-1a). Die Patienten hatten vor ihrem Einschluss in die Studie mindestens 1 Schub unter einer Behandlung mit Beta-Interferon oder Glatirameracetat erlitten, nachdem sie das Arzneimittel zuvor mindestens 6 Monate erhalten hatten (unzureichendes Ansprechen auf vorherige Therapie). Bei Studienbeginn betrug das Durchschnittsalter 35 Jahre, die mittlere Erkrankungsdauer 4,5 Jahre und der mittlere EDSS-Score 2,7. Bei Studienbeginn betrug die mittlere Expositionsdauer mit vorherigen MS-Therapien (≥1 Arzneimittel) in der 12-mg-Lemtrada-Gruppe 35 Monate, und 29% hatten ≥2 vorherige MS-Therapien erhalten.
Die ARR war bei den mit 12 mg Lemtrada behandelten Patienten über den Zeitraum von 2 Jahren im Vergleich zu den mit IFNB-1a behandelten Patienten signifikant um 49% reduziert. Darüber hinaus erzielte die Behandlung mit Lemtrada im Vergleich zu IFNB-1a über 2 Jahre eine signifikante Reduktion des Risikos für eine 6 Monate anhaltende CDW um 42%. Wichtige sekundäre Endpunkte waren die Änderung des EDSS-Scores gegenüber dem Ausgangswert und MRT-Parameter. Bei den mit Lemtrada behandelten Patienten kam es über den Zeitraum von 2 Jahren zu einer signifikanten Reduktion des mittleren EDSS-Scores, während der mittlere EDSS-Score bei den mit IFNB-1a behandelten Patienten signifikant gegenüber dem Ausgangswert anstieg. Die mit Lemtrada behandelten Patienten hatten eine 2,6mal höhere Wahrscheinlichkeit für eine anhaltende bestätigte Verbesserung der Behinderung (CDI für engl. «Confirmed Disability Improvement») als die mit IFNB-1a behandelten Patienten. Der Einfluss der Behandlung auf klinische Endpunkte wurde durch einen signifikanten Effekt auf MRT-Parameter der Entzündung und Krankheitsprogression unterstützt, wie unter anderem das Gehirnvolumen.
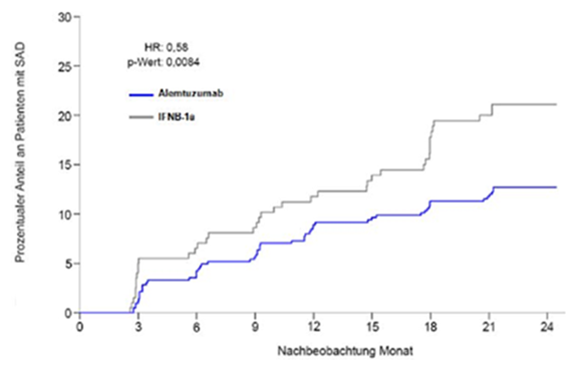
Die Ergebnisse sind in Tabelle 3 und Abbildung 1 dargestellt.
Tabelle 3: Wichtige klinische und MRT-Endpunkte aus Studie 2
|
Endpunkt
|
Lemtrada
(n=426)
|
IFNB-1a s.c.
(n=202)
| |
Klinische Endpunkte
| |
Schubrate (koprimärer Endpunkt)1
| |
Patienten mit irgendeinem Ereignis (Anzahl der Ereignisse)
|
147 (236)
|
104 (201)
| |
ARR (95%-KI)
|
0,26
(0,21; 0,33)
|
0,52
(0,41; 0,66)
| |
Relatives Risiko (95%-KI)
|
0,51
(0,39; 0,65)
|
| |
Risikoreduktion
|
49,40
|
| |
p-Wert
|
<0,0001
|
| |
Behinderung (CDW ≥6 Monate; koprimärer Endpunkt)2
| |
Schätzung der Patienten mit einer 6-monatigen CDW (95%-KI)
|
12,71
(9,89; 16,27)
|
21,13
(15,95; 27,68)
| |
Relatives Risiko (95%-KI)
|
0,58
(0,38; 0,87)
|
| |
p-Wert
|
0,0084
|
| |
Anteil der nach 2 Jahren schubfreien Patienten (%)
| |
Schätzung (95%-KI)
|
65,38
(60,65; 69,70)
|
46,70
(39,53; 53,54)
| |
p-Wert
|
<0,0001
|
| |
Änderung des EDSS gegenüber dem Ausgangswert nach 2 Jahren
| |
(95%-KI)
|
-0,17
(-0,29; -0,05)
|
0,24
(0,07; 0,41)
| |
p-Wert
|
<0,0001
|
| |
Bestätigte Verbesserung der Behinderung (CDI)
| |
Schätzung der Patienten mit einer 6-monatigen CDI (95%-KI)
|
28,82
(24,18; 34,13)
|
12,93
(8,34; 19,77)
| |
Relatives Risiko (95%-KI)
|
2,57
(1,57; 4,20)
|
| |
p-Wert
|
0,0002
|
| |
MRT-Endpunkte
| |
Änderung des T2-Läsionsvolumens gegenüber dem Ausgangswert nach 2 Jahren (%)
|
-1,27
|
-1,23
| |
p-Wert
|
0,1371
|
| |
Patienten mit neuen / sich vergrössernden T2-Läsionen bis Jahr 2 (%)
|
46,2
|
67,9
| |
p-Wert
|
<0,0001
|
| |
Patienten mit Gadolinium-anreichernden Läsionen bis Jahr 2 (%)
|
18,5
|
34,2
| |
p-Wert
|
<0,0001
|
| |
Patienten mit neuen hypointensen T1-Läsionen bis Jahr 2 (%)
|
19,9
|
38,0
| |
p-Wert
|
<0,0001
|
| |
Änderung der Hirnatrophie gegenüber dem Ausgangswert nach 2 Jahren (%)
|
-0,615
|
-0,810
| |
p-Wert
|
0,0121
|
|
Beim EDSS-Score ist die mittlere Änderung angegeben, ermittelt mit einem gemischten Modell für wiederholte Messungen. Beim T2-Läsionsvolumen in der MRT und bei der Hirnatrophie sind die medianen Änderungen angegeben.
1 Koprimärer Endpunkt: ARR und CDW. Die Studie wurde als erfolgreich bewertet, wenn wenigstens einer der beiden koprimären Endpunkte erreicht wurde.
2 Die bestätigte Verschlechterung der Behinderung (CDW) war definiert als Anstieg um mindestens 1 Punkt auf der erweiterten Kurtzke-Skala (Expanded Disability Status Scale, EDSS), ausgehend von einem Baseline-EDSS-Wert von ≥1,0 (Anstieg um 1,5 Punkte bei Patienten mit einem Baseline-EDSS-Wert von 0), über 6 Monate anhaltend.
Abbildung 1: Zeit bis zu einer über 6 Monate anhaltenden bestätigten Verschlechterung der Behinderung in Studie 2
Ergänzende Analysen zeigten, dass Lemtrada 12 mg/Tag im Einklang mit seiner Wirkung auf die Schubrate im Vergleich zu IFNB-1a auch zu einer signifikanten Reduktion der Patienten mit schwerem Schub (Reduktion um 48%; p = 0,0121) und der mit Kortikosteroiden (Reduktion um 56%; p < 0,0001) oder stationär behandelten Schübe (Reduktion um 55%; p = 0,0045) führte.
Die bestätigte Verbesserung der Behinderung ( «Confirmed Disability Improvement», CDI) war definiert als Rückgang um mindestens einen Punkt auf der EDSS ausgehend von einem Baseline-EDSS-Wert von ≥2, der über mindestens 6 Monate anhielt. CDI ist eine Messgrösse für eine anhaltende Verbesserung von Behinderungen. Insgesamt 29% der mit Lemtrada behandelten Patienten erreichten in Studie 2 eine CDI, während nur 13% der mit subkutan verabreichtem IFNB-1a behandelten Patienten diesen Endpunkt erreichten. Der Unterschied war statistisch signifikant (p=0,0002).
Studie 3 (Phase-2-Studie CAMMS223) untersuchte die Sicherheit und Wirksamkeit von Lemtrada bei Patienten mit RRMS über einen Zeitraum von 3 Jahren. Die Patienten wiesen bei ihrem Einschluss in die Studie einen EDSS-Score von 0–3,0, mindestens 2 klinische MS-Episoden in den vorangegangenen 2 Jahren und ≥1 Gadolinium-anreichernde Läsion auf. Sie hatten vor ihrem Einschluss in die Studie noch keine Therapie für ihre MS erhalten. Die Patienten erhielten eine Behandlung mit Lemtrada 12 mg/Tag (n=108) oder 24 mg/Tag (n=108), jeweils als einmal tägliche i.v. Infusion über 5 Tage in Monat 0 und 3 Tage in Monat 12, oder s.c. IFNB-1a 44 µg (n=107) dreimal wöchentlich über 3 Jahre. Sechsundvierzig Patienten erhielten nach 24 Monaten einen dritten 3-tägigen Behandlungszyklus mit Lemtrada 12 mg/Tag oder 24 mg/Tag.
Nach 3 Jahren hatte Lemtrada das Risiko für eine 6-monatige CDW im Vergleich zu IFNB-1a um 76% (relatives Risiko: 0,24 [95%-KI: 0,110; 0,545]; p<0,0006) und die ARR um 67% verringert (relatives Risiko: 0,33 [95%-KI: 0,196; 0,552]; p<0,0001). Alemtuzumab 12 mg/Tag war über einen Beobachtungszeitraum von 2 Jahren mit signifikant niedrigeren Punktwerten im EDSS-Score (Verbesserung gegenüber dem Ausgangswert) verbunden als IFNB-1a (p<0,0001).
In der Subgruppe der RRMS-Patienten, die zu Beginn zwei oder mehr Schübe im Vorjahr und mindestens eine Gd-anreichernde T1-Läsion aufwiesen, betrug die annualisierte Schubrate 0,26 (95%-KI: 0,20; 0,34) in der mit Lemtrada behandelten Gruppe (n=205) und 0,51 (95%-KI: 0,40; 0,64) in der IFNB-1a-Gruppe (n=102) (p<0,0001). Diese Analyse umfasst aufgrund unterschiedlicher MRT-Erfassungsalgorithmen in den Phase-2- und Phase-3-Studien nur Daten aus Phase-3-Studien (CAMMS324 und CAMMS323). Diese Ergebnisse wurden aus einer Post-hoc-Analyse gewonnen und sind mit Vorsicht zu interpretieren.
Nach 5 Jahren hatte Lemtrada das CDW-Risiko im Vergleich zu s.c. IFNB-1a um 69% (Hazard Ratio: 0,31 [95%-KI: 0,161; 0,598]; p=0,0005) und die ARR um 66% (relatives Risiko: 0,34 [95%-KI: 0,202; 0,569]; p<0,0001) verringert.
Studie 4 (CAMMS03409) war eine offene, Bewerter-verblindete, multizentrische Verlängerungsstudie der Phase III zur Langzeitwirksamkeit und -sicherheit von Lemtrada bei Patienten mit RRMS, die zuvor an Studie 1, 2 oder 3 (Phase-III- und -II-Vorgängerstudien) teilgenommen hatten. Die Wirksamkeit und Sicherheit wurden damit über einen medianen Zeitraum von 6 Jahren untersucht, seit Aufnahme der Patienten in Studie 1 und 2 gerechnet.
In Studie 4 konnten die Patienten einen zusätzlichen Behandlungszyklus mit Lemtrada erhalten, wenn sie mindestens eins der folgenden Kriterien erfüllten:
a.hatten mindestens einen klinischen Schub erlitten
b.hatten innerhalb eines Jahres mindestens zwei Einzelläsionen im Gehirn oder Rückenmark laut Bildgebung (MRT) entwickelt, definiert durch die folgenden Aspekte:
·Gadolinium-anreichernde Läsion(en) (generell ≥3 mm in jeder Dimension)
·Neu aufgetretene oder vergrösserte Läsionen in der T2-Gewichtung (generell ≥3 mm in jeder Dimension oder Vergrösserung um ≥3 mm)
Zusätzliche Behandlungszyklen mit Lemtrada wurden in Dosen von 12 mg/Tag an 3 aufeinanderfolgenden Tagen (Gesamtdosis von 36 mg) mindestens 12 Monate nach dem jeweils vorhergehenden Lemtrada-Behandlungszyklus verabreicht.
Von den Patienten, die in Studie 1 und 2 mit Lemtrada 12 mg behandelt wurden, nahmen 91,8% an Studie 4 teil. Hiervon wiederum schlossen 82,7% die Studie vollständig ab. Von den Patienten, die in den Studien 1 und 2 anfänglich mit Lemtrada 12 mg/Tag behandelt und in die Studie 4 aufgenommen worden waren, erhielt ungefähr die Hälfte (51,2%) im gesamten Beobachtungszeitraum von 6 Jahren nur die ersten 2 Lemtrada-Behandlungszyklen und keinerlei weitere krankheitsmodifizierende Therapie.
In den 6 Jahren ab der ersten Lemtrada-Gabe wurden bei den Patienten, die an der Nachbeobachtung teilnahmen, Raten von MS-Schüben, sich entwickelnden Hirnläsionen im MRT und Hirnvolumenverlust ermittelt, die sich mit den Behandlungseffekten von Lemtrada in den Studien 1 und 2 deckten, bei mehrheitlich stabilen oder verbesserten Scores für den Grad der Behinderung. Die Patienten, die anfänglich in Studie 1 und 2 mit Lemtrada behandelt wurden, einschliesslich derer, die an der Nachbeobachtung im Rahmen von Studie 4 teilnahmen, hatten jährliche Schubraten (ARR) von 0,17 bzw. 0,23; hierbei hatten 22,3% bzw. 29,7% der Patienten aus Studie 1 und 2 eine bestätigte Verschlechterung der Behinderung (CDW) und 32,7% und 42,5% eine bestätigte Verbesserung der Behinderung (CDI). Bei den Patienten, die einen oder mehrere zusätzliche Lemtrada-Behandlungszyklen erhalten hatten, waren im Vergleich zum jeweiligen Vorjahr Verbesserungen der Schubrate, der MRT-Aktivität und der mittleren Behinderungs-Scores nach einer ersten oder zweiten zusätzlichen Behandlung (Zyklus 3 bzw. 4) zu verzeichnen. Da jedoch die Studie 4 offen und nicht-kontrolliert angelegt war, sind die Ergebnisse dieser Studie nicht geeignet, einen definitiven Nachweis der Wirksamkeit zu erbringen.
Nutzen und Risiken von 5 oder mehr Behandlungszyklen wurden bisher nicht untersucht.
Immunogenität
Wie bei allen therapeutisch verabreichten Proteinen besteht ein Potential für immunogene Wirkungen. Die Daten spiegeln den prozentualen Anteil der Patienten wider, deren Untersuchungsergebnisse als positiv für Anti-Alemtuzumab-Antikörper bewertet wurden (gemessen mittels Enzyme-linked-immumosorbent-Assay [ELISA] und bestätigt mittels kompetitivem Bindungsassay). Positiv getestete Proben wurden mittels Durchflusszytometrie weiter auf Anzeichen einer In-vitro-Hemmung untersucht. In den kontrollierten klinischen Studien zur MS erfolgten Blutentnahmen für die Bestimmung von Anti-Alemtuzumab-Antikörpern im Serum 1, 3 und 12 Monate nach jedem Behandlungszyklus. Etwa 85% der mit Lemtrada behandelten Patienten wurden im Verlauf der Studie positiv auf Anti-Alemtuzumab-Antikörper getestet und > 90% dieser Patienten wurden auch positiv auf Antikörper getestet, die in vitro die Bindung von Lemtrada hemmten. Wenn Anti-Alemtuzumab-Antikörper nachweisbar waren, so bildeten sich diese bis 15 Monate nach der ersten Exposition aus. Während zwei Behandlungszyklen bestand kein Zusammenhang zwischen dem Vorliegen von Anti-Alemtuzumab-Antikörpern bzw. hemmenden Anti-Alemtuzumab-Antikörpern und einer verminderten Wirksamkeit oder dem Auftreten von unerwünschten Arzneimittelwirkungen einschliesslich von infusionsassoziierten Reaktionen. Hohe Titer von Anti-Alemtuzumab-Antikörpern wurden bei einigen Patienten beobachtet und waren mit einer unvollständigen Lymphozytendepletion im Anschluss an einen dritten oder vierten Behandlungszyklus assoziiert. Es waren jedoch keine Auswirkungen der Anti-Alemtuzumab-Antikörper auf die klinische Wirksamkeit oder das Sicherheitsprofil von Lemtrada zu erkennen.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Lemtrada bei Kindern mit MS im Alter von 0 bis 18 Jahren ist nicht erwiesen. Lemtrada ist nicht für die Anwendung bei Kindern und Jugendlichen indiziert (siehe «Kontraindikationen»).
|