Eigenschaften/WirkungenATC-Code
L01FF03
Wirkungsmechanismus / Pharmakodynamik
Die Expression des programmierten Zelltod-Liganden-1 (PD-L1) ist eine adaptive Immunantwort, die Tumore vor dem Immunsystem verbirgt und beschützt. PD-L1 kann durch Entzündungssignale (z.B. IFN-gamma) induziert werden und sowohl auf Tumorzellen als auch auf tumorassoziierten Immunzellen in einer Tumor-Mikroumgebung exprimiert werden. PD-L1 blockiert die Funktion der T-Zellen und deren Aktivierung aufgrund der Interaktion mit PD-1 und CD80 (B7-1). Durch Andocken an ihre Rezeptoren verringert PD-L1 die Aktivität, Vermehrung und Zytokin-Produktion von T-Zellen.
Durvalumab ist ein vollständig humaner, stark bindungsaffiner, monoklonaler Immunglobulin-G1-kappa-Antikörper (IgG1κ), der die Interaktion von PD-L1 mit PD-1 und CD80 (B7-1) selektiv blockiert, dabei jedoch die PD-1/PD-L2-Interaktion intakt lässt. Durvalumab ruft keine antikörperabhängige zellvermittelte Zytotoxizität (ADCC) hervor. Das selektive Blockieren der PD-L1/PD-1- und PD-L1/CD80-Interaktionen steigert die Antitumor-Immunantworten. Diese Antitumor-Antworten können die Eliminierung von Tumoren bewirken.
In präklinischen Studien führte die PD-L1-Blockade zu erhöhter T-Zellen-Aktivierung und verringerten Tumorgrössen.
Klinische Wirksamkeit
Nicht-kleinzelliges Lungenkarzinom (NSCLC) – PACIFIC Studie
Basierend auf der modellierten Beziehung zwischen Exposition und Wirksamkeit/Sicherheit, unterstützt durch limitierte klinische Daten, ergeben sich für die Imfinzi Dosierungen von 10 mg/kg alle 2 Wochen und 1500 mg alle 4 Wochen bei Patienten mit lokal fortgeschrittenem, nicht resezierbarem nicht-kleinzelligem Lungenkarzinom (NSCLC), deren Erkrankung nach einer definitiven platinbasierten Chemoradiotherapie nicht fortgeschritten ist keine klinisch relevanten Unterschiede.
Die Wirksamkeit von Imfinzi wurde in der PACIFIC-Studie ermittelt, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Studie mit 713 Patienten mit histologisch oder zytologisch nachgewiesenem, lokal fortgeschrittenem, inoperablem NSCLC. Die Patienten hatten innerhalb eines Zeitraums von 1 bis 42 Tagen vor Beginn der Studienbehandlung mindestens zwei Zyklen einer definitiven platinbasierten Radiochemotherapie abgeschlossen und verfügten über einen ECOG-Leistungsstatus von 0 oder 1. 92 % der Patienten hatten eine Gesamtdosis von 54 bis 66 Gy Strahlung erhalten. Nicht für diese Studie infrage kamen Patienten, die nach einer Radiochemotherapie eine Progression aufwiesen, Patienten mit aktiver oder zuvor dokumentierter Autoimmunerkrankung innerhalb eines Zeitraums von 2 Jahren vor Studienbeginn, einer vorbestehenden Immunschwäche, vorbestehenden schweren immunvermittelten unerwünschten Reaktionen, Erkrankungen, die eine systemische Immunsuppression notwendig machten ausgenommen eine physiologische Dosis systemischer Kortikosteroide, Patienten mit aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion oder Patienten, die innerhalb von 30 Tagen vor oder nach dem Beginn der Behandlung mit Imfinzi attenuierten Lebendimpfstoff erhielten. Die Patienten wurden 2:1 randomisiert und erhielten 12 Monate lang oder bis zu inakzeptabler Toxizität oder bis zu bestätigter Krankheitsprogression alle 2 Wochen entweder 10 mg/kg Imfinzi (n=476) oder Placebo (n=237) mittels intravenöser Infusion. Die Randomisierung wurde stratifiziert nach Geschlecht, Alter (< 65 Jahre vs. ≥65 Jahre) und Raucherstatus (Raucher vs. Nichtraucher). Tumorbeurteilungen wurden während den ersten 12 Monaten alle 8 Wochen und danach alle 12 Wochen durchgeführt.
Die Patienten wurden unabhängig von ihrem Tumor-PD-L1-Expressionsstatus in die Studie eingeschlossen. Soweit verfügbar, wurden archivierte Tumorgewebeproben, die vor der Radiochemotherapie entnommen wurden, mit dem VENTANA PD-L1 (SP263) IHC-Assay retrospektiv auf PD-L1-Expression auf Tumorzellen (TC) getestet.
Die demografischen und krankheitsbedingten Eigenschaften bei Studienbeginn waren zwischen den Studienarmen ausgewogen. Die Zusammensetzung der gesamten Studienpopulation zu Studienbeginn war wie folgt: Männer (70 %), Alter ≥65 Jahre (45 %), hellhäutig (69 %), asiatisch (27 %), sonstige (4 %), Raucher (16 %), ehem. Raucher (75 %) und nie geraucht (9 %), WHO/ECOG PS 0 (49 %), WHO/ECOG PS 1 (51 %). Die krankheitsbedingten Eigenschaften waren wie folgt: Stadium IIIA (53 %), Stadium IIIB (45 %), histologische squamöse Untergruppe (46 %), nicht-squamös (54 %).
Die zwei primären Endpunkte der Studie waren Gesamtüberleben (OS) und progressionsfreies Überleben (PFS) von Imfinzi vs. Placebo.
Die Studie zeigte eine statistisch signifikante Verbesserung des OS in der Imfinzi-Gruppe verglichen mit der Placebo-Gruppe [Hazard-Ratio (HR) = 0,68 (95 %-KI: 0,53; 0,87), p = 0,00251]. Zum Zeitpunkt der vorläufigen OS-Analyse (22. März 2018) betrug die mediane Nachbeobachtungszeit 25,2 Monate; 299 Patienten (183 (38,4 %) in der Durvalumab-Gruppe und 116 (48,9 %) in der Placebo-Gruppe) waren verstorben. Das mediane Gesamtüberleben wurde in der Durvalumab-Gruppe noch nicht erreicht; in der Placebo-Gruppe war es gemäss Kaplan-Meier Schätzung 28,7 Monate. Das Gesamtüberleben nach 24 Monaten betrug 66,3 % in der Imfinzi-Gruppe und 55,6 % in der Placebo-Gruppe.
Zum Zeitpunkt der Aktualisierung der Daten nach der 5-jährigen Nachbeobachtung waren insgesamt 23 neue OS-Ereignisse seit der zweiten Analyse der OS-Nachbeobachtung gemeldet worden (insgesamt 419 Todesfälle [264 in der Imfinzi-Gruppe und 155 in der Placebo-Gruppe]). Der Überlebensnutzen von Imfinzi gegenüber Placebo war konsistent mit der primären OS-Analyse, bei einer Reduktion des Mortalitätsrisikos gegenüber Placebo um 28 % [HR = 0,72 (95%-KI: 0,59-0,89)]. Die Kaplan-Meier-Schätzung des medianen OS betrug 29,1 Monate in der Placebo-Gruppe gegenüber 47,5 Monaten in der Imfinzi-Gruppe. Die geschätzten Gesamtüberlebensraten lagen bei 42,9 % für die Imfinzi-Gruppe und 33,4 % für die Placebo-Gruppe.
Zum Zeitpunkt der Interimsanalyse (13. Februar 2017) durch einen verblindeten, unabhängigen, zentralen Review (BICR) gemäss den RECIST 1.1 Kriterien zeigte die Studie eine statistisch signifikante Verbesserung des PFS in der Imfinzi-Gruppe verglichen mit der Placebo-Gruppe [HR = 0,52 (95 %-KI: 0,42; 0,65), p < 0,0001]. Zu diesem Zeitpunkt waren 371 Patienten (214 (45,0 %) in der Durvalumab-Gruppe und 157 (66,2 %) in der Placebo-Gruppe) progredient oder verstorben. Das mediane PFS gemäss Kaplan-Meier Schätzung war 16,8 Monate in der Imfinzi-Gruppe und 5,6 Monate in der Placebo-Gruppe. Das PFS nach 12 Monaten betrug 55,9 % in der Imfinzi-Gruppe und 35,5 % in der Placebo-Gruppe. Das PFS nach 18 Monaten betrug 44,2 % in der Imfinzi-Gruppe und 27,0 % in der Placebo-Gruppe. Die Verbesserungen des OS und des PFS zugunsten der Patienten in der Imfinzi-Gruppe im Vergleich zur Placebo-Gruppe wurde durchgehend in allen analysierten vordefinierten Subgruppen (Ethnizität, Alter, Geschlecht, Raucherstatus, EGFR Mutationsstatus und Histologie) beobachtet.
Zum Zeitpunkt der Analyse der 5-jährigen Nachbeobachtung (11. Januar 2021) basierend auf den BICR-Bewertungen des PFS nach RECIST 1.1 war der in der Imfinzi-Gruppe festgestellte PFS-Nutzen [HR: 0,55 (95%-KI: 0,45-0,68)] konsistent mit der primären Analyse des PFS [HR: 0,52; (95%-KI: 0,42-0,65)]. Die Kaplan-Meier-Schätzung des medianen PFS betrug 16,9 Monate in der Durvalumab-Gruppe (95%-KI: 13,0-23,9) im Vergleich zu 5,6 Monaten in der Placebo-Gruppe (95%-KI: 4,8-7,7).
In der Imfinzi-Gruppe trat bei 21 (4,4 %) Patienten eine unerwünschte Wirkung mit Todesfolge auf, in der Placebo-Gruppe trat bei 14 (6,0 %) Patienten eine unerwünschte Wirkung mit Todesfolge auf.
Post-hoc explorative Subgruppenanalyse entsprechend der PD-L1-Expression
Von den 713 randomisierten Patienten der PACIFIC-Studie waren 451 (63 %) der Gewebeproben von ausreichender Qualität und Quantität zur Bestimmung der PD-L1-Expression. Von den 451 Proben hatten 33 % einen PD-L1 TC < 1 %, 67 % PD-L1 TC ≥1 %, 32 % PD-L1 TC 1 - < 25 %, 35 % PD-L1 TC ≥25 %.
Post hoc Subgruppenanalysen wurden durchgeführt, um die Wirksamkeit bei zusätzlichen, nicht im statistischen Analyseplan vorgesehenen PD-L1-Expressionsleveln des Tumors (< 1 %, ≥1 %, 1 bis < 25 %) zu bewerten.
PFS:
In der Subgruppe mit fehlender PD-L1 Expression (PD-L1 TC < 1 %) war die PFS-HR 0,73; 95 %-KI: 0,48, 1,11.
Für die untersuchten Subgruppen mit positiver PD-L1 Expression (PD-L1 TC ≥1%) und der Subgruppe mit nicht evaluierbarer PD-L1 Expression (PD-L1 TC unbekannt) wurden folgende Resultate ermittelt: PD-L1 TC ≥1 % HR: 0,46; 95 %-KI: 0,33, 0,64; PD-L1 TC 1 bis < 25 % HR: 0,49; 95 %-KI:0,30, 0,80; PD-L1 TC ≥25 % HR: 0,41; 95 %-KI: 0,26, 0,65; PD-L1 TC unbekannt HR: 0,59; 95 %-KI: 0,42, 0,83).
OS:
In der Subgruppe mit fehlender PD-L1 Expression (PD-L1 TC < 1 %) war die OS-HR 1.36; 95 %-KI: 0,79, 2,34.
Für die untersuchten Subgruppen mit positiver PD-L1 Expression (PD-L1 TC ≥1%) und mit nicht evaluierbarer PD-L1 Expression (PD-L1 TC unbekannt) wurden folgende Resultate ermittelt: PD-L1 TC ≥1 % HR: 0,53; 95 %-KI: 0,36, 0,77; PD-L1 TC 1 bis < 25 % HR: 0,60; 95 %-KI: 0,35, 1,03; PD-L1 TC ≥25 % HR: 0,46; 95 %-KI: 0,27, 0,78; PD-L1 TC unbekannt HR: 0,62; 95 %-KI: 0,43, 0,89.
Resezierbares NSCLC – AEGEAN-Studie
AEGEAN ist eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Phase-III-Studie zur Beurteilung der Wirksamkeit von Imfinzi in Kombination mit Chemotherapie als neoadjuvante Behandlung, gefolgt von einer Imfinzi-Monotherapie nach der Operation, bei Patienten mit resezierbarem NSCLC (Stadium IIA bis ausgewählte Patienten mit Stadium IIIB (N2) [AJCC, 8. Auflage]). In die Studie wurden zuvor unbehandelte Patienten mit dokumentiertem plattenepithelialem oder nicht plattenepithelialem NSCLC ohne vorherige Exposition gegenüber einer Immuntherapie, mit einem WHO/ECOG Performance Status von 0 oder 1 und mindestens einer Zielläsion nach RECIST 1.1 aufgenommen. Vor der Randomisierung wurde der PD-L1-Expressionsstatus des Tumors mit dem Ventana PD-L1 (SP263) Test bestätigt.
Patienten mit aktiver oder zuvor dokumentierter Autoimmunerkrankung oder Anwendung einer immunsuppressiven Medikation innerhalb von 14 Tagen vor der ersten Dosis von Durvalumab wurden aus der Studie ausgeschlossen. Aus der Studie ausgeschlossen waren Patienten, deren geplante Operation eine Segmentresektion, eine Keilresektion oder (nach einer Prüfplanänderung) eine Pneumektomie war. Während der Studie durchgeführte Operationen wurden im Rahmen der Standardtherapie durchgeführt und konnten eine Pneumektomie umfassen. Nach einer Prüfplanänderung wurden später keine Patienten mit T4-Tumoren mit Invasion von Zwerchfell, Mediastinum, Herz, grossen Gefässen, Trachea, Nervus laryngeus recurrens, Ösophagus oder Carina mehr in die Studie aufgenommen. Aus der Studienpopulation für die Wirksamkeitsanalyse (modifizierte Intent-to-treat-Analyse [mITT]) wurden Patienten mit bekannten EGFR-Mutationen oder ALK-Rearrangements ausgeschlossen.
Die Randomisierung erfolgte stratifiziert nach Krankheitsstadium (Stadium II vs. Stadium III) und PD-L1-Expressionsstatus (TC < 1 % vs. TC ≥1 %).
In der AEGEAN-Studie wurden 802 Patienten im Verhältnis 1:1 für die perioperative Behandlung mit Imfinzi (Arm 1) oder Placebo (Arm 2) in Kombination mit neoadjuvanter Chemotherapie randomisiert. Ein Crossover zwischen den Studienarmen war nicht erlaubt. Die Wirksamkeitsanalyse wurde auf der Basis von 740 Patienten in der mITT-Population durchgeführt.
·Arm 1: Imfinzi 1500 mg + Chemotherapie alle 3 Wochen für bis zu 4 Zyklen vor der Operation, gefolgt von Imfinzi 1500 mg alle 4 Wochen für bis zu 12 Zyklen nach der Operation.
·Arm 2: Placebo + Chemotherapie alle 3 Wochen für bis zu 4 Zyklen vor der Operation, gefolgt von Placebo alle 4 Wochen für bis zu 12 Zyklen nach der Operation.
Die Wahl der Standardchemotherapie lag unter Berücksichtigung der Histologie im Ermessen des Prüfarztes. Bei plattenepithelialen Tumoren: Carboplatin (AUC 6) und Paclitaxel (200 mg/m2) an Tag 1 jedes 3-wöchigen Zyklus für maximal 4 Zyklen oder Cisplatin (75 mg/m2) an Tag 1 und Gemcitabin (1250 mg/m2) an den Tagen 1 und 8 jedes 3-wöchigen Zyklus für maximal 4 Zyklen. Die Anwendung von Carboplatin (AUC 5) kombiniert mit Gemcitabin (anstatt Cisplatin) war bei Patienten erlaubt, die Begleiterkrankungen hatten oder Cisplatin nach Einschätzung des Prüfarztes nicht vertrugen. Bei nicht plattenepithelialen Tumoren: Pemetrexed (500 mg/m2) und Cisplatin (75 mg/m2) an Tag 1 jedes 3-wöchigen Zyklus für maximal 4 Zyklen oder Pemetrexed (500 mg/m2) und Carboplatin (AUC 5) an Tag 1 jedes 3-wöchigen Zyklus für maximal 4 Zyklen. Bei ungünstiger Verträglichkeit konnten die Patienten an jedem Punkt der Studie von der Cisplatin- zur Carboplatin-Therapie wechseln (wenn sie für diese Therapie in Frage kamen).
Patienten, die sich einer Operation mit R0/R1-Resektionsrändern unterzogen hatten, konnten die adjuvante Behandlung fortsetzen.
Eine Tumorbeurteilung nach RECIST 1.1 wurde zur Baseline und nach Abschluss der neoadjuvanten Phase (vor der Operation) durchgeführt. Die erste postoperative CT-/MRT-Untersuchung von Thorax und Abdomen (einschliesslich der gesamten Leber und beider Nebennieren) wurde 5 Wochen ±2 Wochen nach der Operation und vor, aber möglichst nahe am Beginn der adjuvanten Therapie, durchgeführt. Anschliessend erfolgten Tumorbeurteilungen alle 12 Wochen (bezogen auf das Operationsdatum) bis Woche 48, alle 24 Wochen (bezogen auf das Operationsdatum) bis Woche 192 (ca. 4 Jahre) und danach alle 48 Wochen (bezogen auf das Operationsdatum) bis zur radiologisch bestätigten Krankheitsprogression (Progressive disease; PD) gemäss RECIST 1.1, bis zum Widerruf der Einwilligung oder bis zum Tod.
Der Überlebensstatus wurde 2, 3 und 4 Monate nach Beendigung der Behandlung, anschliessend alle 2 Monate bis Monat 12 und danach alle 3 Monate überprüft.
Die primären Endpunkte der Studie waren das vollständige pathologische Ansprechen (pathological complete response; pCR) gemäss verblindeter zentraler pathologischer Untersuchung sowie das ereignisfreie Überleben (event-free survival; EFS) gemäss verblindeter unabhängiger zentraler Beurteilung (BICR). Die wichtigsten sekundären Endpunkte waren das bedeutsame pathologische Ansprechen (major pathological response; MPR) gemäss verblindeter zentraler pathologischer Untersuchung, das krankheitsfreie Überleben (disease free survival, DFS) gemäss BICR und Gesamtüberleben (overall survival; OS). Weitere sekundäre Wirksamkeitsziele waren das EFS (Analysepopulation PD-L1-TC ≥1 %) und pCR (Analysepopulation PD-L1-TC ≥1%).
Bei der geplanten Zwischenanalyse des pCR erreichte die Studie die vorgegebene Grenze für die Erklärung der statistischen Signifikanz für pCR und MPR. In der Folgezeit, bei der ersten geplanten Zwischenanalyse des EFS, erreichte die Studie ihre vorgegebene Grenze für die Erklärung der statistischen Signifikanz für das EFS.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren in den beiden Studienarmen ausgewogen (366 Patienten in Arm 1 und 374 Patienten in Arm 2 der mITT-Population). Die demographischen und krankheitsspezifischen Ausgangsmerkmale der Population für die Wirksamkeitsanalyse (mITT) waren wie folgt: männlich (71,6 %), weiblich (28,4 %), Alter ≥65 Jahre (51,6 %), medianes Alter 65 Jahre (Bereich: 30 bis 88 Jahre), WHO/ECOG PS 0 (68,4 %), WHO/ECOG PS 1 (31,6 %), weiss (53,6 %), asiatisch (41,5 %), schwarz bzw. afroamerikanisch (0,9 %), indigen amerikanisch oder indigen alaskisch (1,4 %), andere Abstammung (2,6 %), hispanisch oder lateinamerikanisch (16,1 %), nicht-hispanisch oder -lateinamerikanisch (83,9 %), aktueller oder ehemaliger Raucher (85,5 %), Nie-Raucher (14,5 %), Plattenepithelhistologie (48,6 %) und Nicht-Plattenepithelhistologie (50,7 %), Stadium II (28,4 %), Stadium III (71,6 %), PD-L1-Expressionsstatus TC ≥1 % (66,6 %), PD-L1-Expressionsstatus TC < 1 % (33,4 %). Die demographischen Daten und Ausgangsmerkmale der mITT-Population waren vergleichbar mit der ITT-Population, mit Ausnahme des Fehlens von Patienten mit bekannten EGFR-Mutationen oder ALK-Rearrangements.
Die Studie zeigte eine statistisch signifikante Verbesserung des EFS [HR = 0,68 (95%-KI: 0,53; 0,88), p = 0,003902] im Imfinzi-Arm verglichen mit dem Placebo-Arm. Ausserdem zeigte die Studie eine statistisch signifikante Verbesserung des pCR [Differenz der Anteile 12,96 % (95%-KI: 8,67, 17,57)] im Imfinzi-Arm verglichen mit dem Placebo-Arm. Die Daten zum Gesamtüberleben (OS) hatten keine ausreichende Reife zum Zeitpunkt der EFS-Analyse. Siehe Tabelle 3.
Tabelle 3: Wirksamkeitsergebnisse der AEGEAN-Studie (mITT)
|
|
Imfinzi + Chemotherapie
(n = 366)
|
Placebo + Chemotherapie
(n = 374)
| |
EFSa
| |
Anzahl der Ereignisse, n (%)
|
98 (26,8)
|
138 (36,9)
| |
Medianes EFS (95%-KI) (Monate)
|
NR (31,9; NR)
|
25,9 (18,9; NR)
| |
EFS nach 12 Monaten, % (95%-KI)
|
73,4 (67,9; 78,1)
|
64,5 (58,8; 69,6)
| |
EFS nach 24 Monaten, % (95%-KI)
|
63,3 (56,1; 69,6)
|
52,4 (45,4; 59,0)
| |
Hazard Ratio (95%-KI)
|
0,68 (0,53; 0,88)
| |
2-seitiger p-Wertd
|
0,003902
| |
pCRa,b,d
| |
Anzahl der Patienten mit Ansprechen
|
63
|
16
| |
Ansprechrate, % (95%-KI)
|
17,21 (13,49; 21,48)
|
4,28 (2,46; 6,85)
| |
Differenz der Anteile, % (95%-KI)
|
12,96 (8,67; 17,57)
| |
MPRa,c,d
| |
Anzahl der Patienten mit Ansprechen
|
122
|
46
| |
Ansprechrate, % (95%-KI)
|
33,33 (28,52; 38,42)
|
12,30 (9,15; 16,06)
| |
Differenz der Anteile, % (95%-KI)
|
21,03 (15,14; 26,93)
|
a Die Ergebnisse basieren auf der vorab geplanten EFS-Zwischenanalyse und der abschliessenden pCR-/MPR-Analyse (DCO: 10. November 2022), die 46,3 Monate nach Studienbeginn erfolgte.
b Basierend auf einer vorgegebenen pCR-Zwischenanalyse (DCO: 14. Januar 2022) bei n = 402 war die pCR-Rate statistisch signifikant (p = 0,000036) verglichen mit dem Signifikanzniveau von 0,0082 %.
c Basierend auf einer vorgegebenen MPR-Zwischenanalyse (DCO: 14. Januar 2022) bei n = 402 war die MPR-Rate statistisch signifikant (p = 0,000002) verglichen mit dem Signifikanzniveau von 0,0082 %.
d Der 2-seitige p-Wert für pCR und MPR wurde basierend auf einem stratifizierten CMH-Test berechnet. Der 2-seitige p-Wert für das EFS wurde basierend auf einem stratifizierten Log-Rank-Test berechnet. Die Stratifizierungsfaktoren waren PD-L1 und Krankheitsstadium.
Die Grenze für die Erklärung der statistischen Signifikanz für jeden der Wirksamkeitsendpunkte wurde mit einer Alpha-Spending-Funktion nach Lan-DeMets, die einen O'Brien-Fleming-Ansatz approximiert, bestimmt (EFS = 0,9899 %, pCR = 0,0082 %, MPR = 0,0082 %, 2-seitig).
Zum Zeitpunkt des DCO am 14. August 2023 betrug die Reife in Bezug auf das Gesamtüberleben (OS) 28,6 % (212 OS Ereignisse). In der Subgruppe PD-L1 TC < 1 % der mITT-Population waren 27 Patienten (22,1 %) im Imfinzi + Chemotherapie-Arm und 39 Patienten (31,2 %) im Placebo + Chemotherapie-Arm verstorben. In der Subgruppe PD-L1 TC ≥1 % waren 72 (29,5 %) und 74 (29,7 %) Patienten im jeweiligen Studienarm verstorben.
Kleinzelliges Lungenkarzinom im lokal begrenzten Stadium (LS-SCLC) – ADRIATIC-Studie
In der ADRIATIC-Studie wurde die Wirksamkeit von Imfinzi mit oder ohne Tremelimumab untersucht. ADRIATIC war eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Studie an 730 Patienten mit histologisch oder zytologisch bestätigtem LS-SCLC (Stadium I bis III gemäss AJCC, 8. Ausgabe), deren Erkrankung nach einer gleichzeitigen Chemoradiotherapie nicht fortgeschritten war. Patienten im Stadium I oder II mussten gemäss Feststellung des Prüfarztes internistisch inoperabel sein. Die Patienten schlossen 3-4 Zyklen definitiver platinbasierter Chemoradiotherapie, 60-66 Gy einmal täglich (QD) über 6 Wochen oder 45 Gy zweimal täglich (BID) über 3 Wochen, innerhalb von 1 bis 42 Tagen vor der ersten Dosis der Studienbehandlung ab. Eine prophylaktische Schädelbestrahlung (PCI) konnte im Ermessen des Prüfarztes nach der Chemoradiotherapie innerhalb von 1 bis 42 Tagen vor der ersten Dosis der Studienbehandlung durchgeführt werden.
Von einer Teilnahme ausgeschlossen wurden Patienten mit aktiver oder früherer dokumentierter Autoimmunerkrankung in den letzten 5 Jahren vor Beginn der Studie, anamnestisch bekannter aktiver primärer Immundefizienz, anamnestisch bekannter Pneumonitis Grad ≥2 oder aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion, Patienten mit aktiver interstitieller Lungenerkrankung, Patienten, die innerhalb von 30 Tagen vor Beginn der Behandlung mit Imfinzi attenuierten Lebendimpfstoff erhalten haben sowie Patienten mit Hirnmetastasen, Kompression der Wirbelsäule oder einer Vorgeschichte von leptomeningealer Karzinomatose. Patienten mit gemischter SCLC- und NSCLC-Histologie wurden ebenfalls von einer Teilnahme an der Studie ausgeschlossen.
Die Randomisierung erfolgte stratifiziert nach Stadium (I/II oder III) und PCI (ja oder nein). Die Patienten wurden im Verhältnis 1:1:1 randomisiert und den folgenden Behandlungen zugeteilt:
·Arm 1: Imfinzi 1500 mg + Placebo alle 4 Wochen über 4 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen.
·Arm 2: Placebo + zweites Placebo alle 4 Wochen über 4 Zyklen, gefolgt von einem Placebo alle 4 Wochen.
·Arm 3: Imfinzi 1500 mg + Tremelimumab 75 mg alle 4 Wochen über 4 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen.
Nachdem insgesamt 600 Patienten für die drei Studienarme randomisiert worden waren, wurden nachfolgende Patienten im Verhältnis 1:1 randomisiert Arm 1 oder 2 zugeteilt und erhielten entweder Imfinzi 1500 mg alle 4 Wochen oder Placebo alle 4 Wochen.
Die Behandlung wurde bis zur Krankheitsprogression, inakzeptablen Toxizität oder für maximal 24 Monate fortgesetzt. Tumorbeurteilungen wurden während der ersten 72 Wochen alle 8 Wochen, dann bis zur 96. Woche alle 12 Wochen und danach alle 24 Wochen durchgeführt.
Die demografischen und krankheitsbedingten Eigenschaften bei Studienbeginn waren zwischen den Studienarmen gut ausgewogen. Die demografischen Ausgangsdaten und die Krankheitsmerkmale bei Studienbeginn in der Imfinzi- und der Placebo-Gruppe waren: männlich (69,1 %), Alter ≥65 Jahre (39,2 %), weiss (50,4 %), schwarz oder afroamerikanisch (0,8 %), asiatisch (47,5 %), sonstige (1,3 %), hispanisch oder lateinamerikanisch (4,2 %), aktuelle Raucher (22,3 %), frühere Raucher (68,5 %), nie geraucht (9,2 %), WHO/ECOG-PS 0 (48,7 %), WHO/ECOG-PS 1 (51,3 %), Stadium I (3,6 %), Stadium II (9,1 %), Stadium III (87,4 %).
Vor der Randomisierung erhielten alle Patienten eine platinbasierte Chemotherapie (66,2 % Cisplatin-Etoposid, 33,8 % Carboplatin-Etoposid); 72,1 % der Patienten erhielten eine RT QD (darunter 92,4 % mit ≥60 - ≤66 Gy QD); 27,9 % erhielten eine RT BID (darunter 96,6 % mit 45 Gy BID) und 53,8 % der Patiente erhielten eine PCI. Das Ansprechen auf die CRT war wie folgt: vollständiges Ansprechen (12,3 %), partielles Ansprechen (73,8 %), stabile Erkrankung (14,0 %).
Die zwei primären Endpunkte der Studie waren OS und PFS unter Imfinzi vs. Placebo. Sekundäre Endpunkte waren ORR unter Imfinzi vs. Placebo. Die Beurteilung des PFS und der ORR erfolgte durch BICR gemäss RECIST v1.1.
Bei einer geplanten Zwischenanalyse zeigte die Studie eine statistisch signifikante und klinisch bedeutsame Verbesserung des OS für Imfinzi verglichen mit Placebo [HR=0,73 (95%-KI: 0,569, 0,928), p=0,01042]. Die Studie zeigte ausserdem eine statistisch signifikante und klinisch relevante Verbesserung des PFS für Imfinzi verglichen mit Placebo [HR=0,76 (95%-KI: 0,606, 0,950), p=0,01608]. Siehe Tabelle 4.
Tabelle 4. Wirksamkeitsergebnisse der ADRIATIC-Studie
|
|
Arm 1: Imfinzi (n = 264)
|
Arm 2: Placebo (n = 266)
| |
OSa
| |
Anzahl der Todesfälle (%)
|
115 (43,6)
|
146 (54,9)
| |
Medianes OS (Monate) (95%-KI)b
|
55,9 (37,3, NR)
|
33,4 (25,5, 39,9)
| |
HR (95%-KI)c
|
0,73 (0,569, 0,928)
| |
p-Wertd
|
0,01042
| |
OS nach 24 Monaten (%) (95%-KI)b
|
68,0 (61,9, 73,3)
|
58,5 (52,3, 64,3)
| |
OS nach 36 Monaten (%) (95%-KI)b
|
56,5 (50,0, 62,5)
|
47,6 (41,3, 53,7)
| |
PFSe
| |
Anzahl der Ereignisse (%)
|
139 (52,7)
|
169 (63,5)
| |
Medianes PFS (Monate) (95%-KI)b
|
16,6 (10,2, 28,2)
|
9,2 (7,4, 12,9)
| |
HR (95%-KI)f
|
0,76 (0,606, 0,950)
| |
p-Wertd
|
0,01608
| |
PFS nach 18 Monaten (%) (95%-KI)b
|
48,8 (42,2, 55,0)
|
36,1 (29,9, 42,2)
| |
PFS nach 24 Monaten (%) (95%-KI)b
|
46,2 (39,6, 52,5)
|
34,2 (28,2, 40,3)
| |
ORRe
| |
ORRg n (%)
|
53/175 (30,3)
|
54/169 (32,0)
| |
Vollständiges Ansprechen n (%)
|
5 (2,9)
|
4 (2,4)
| |
Partielles Ansprechen n (%)
|
48 (27,4)
|
50 (29,6)
| |
Odds Ratio (95%-KI)
|
-1,2 (-11,0, 8,5)
| |
Mediane DoRb,e (Monate)
(95%-KI)
|
33,0 (22,4, NR)
|
27,7 (9,6, NR)
| |
Anteil an Patienten in Remission nach 12 Monatenb,e (%) (95%-KI)
|
73,7 (59,0, 83,8)
|
60,3 (44,5, 72,9)
| |
Anteil an Patienten in Remission nach 18 Monatenb,e (%) (95%-KI)
|
71,5 (56,6, 82,0)
|
55,2 (39,4, 68,5)
|
a Die mediane Dauer der OS-Nachbeobachtung bei zensierten Patienten betrug 37,19 Monate in der Imfinzi-Gruppe und 37,24 Monate in der Placebo-Gruppe.
b Mittels Kaplan-Meier-Methode berechnet. KI für den Median mit der Brookmeyer-Crowley-Methode generiert.
c Die Analyse für HR erfolgte mit einem stratifizierten Cox-Proportional-Hazards-Modell und der 2-seitige p-Wert basiert auf einem stratifizierten Log-Rank-Test; beide sind für Behandlung mit PCI adjustiert.
d p-Wert basierend auf den Ergebnissen der vorab geplanten Zwischenanalyse. Basierend auf einer Lan-DeMets-Alpha-Spending-Funktion mit O'Brien-Fleming-Grenze und der tatsächlich beobachteten Anzahl der Ereignisse lag die Grenze für die Erklärung der statistischen Signifikanz für OS bei 0,01679 für ein Gesamt-Alpha von 4,5 % und für PFS bei 0,02805 für ein Gesamt-Alpha von 5 % (Lan und DeMets, 1983).
e Beurteilt vom BICR gemäss RECIST v1.1.
f Die Analyse für HR erfolgte mit einem stratifizierten Cox-Proportional-Hazards-Modell und der 2-seitige p-Wert basiert auf einem stratifizierten Log-Rank-Test; beide sind für TNM-Stadium und Behandlung mit PCI adjustiert.
g Basierend auf Subgruppe des vollständigen Analysesets mit messbarer Erkrankung zur Baseline gemäss RECIST v1.1; Imfinzi (n = 175), Placebo (n = 169).
Die Verbesserungen von OS und PFS zugunsten der Patienten, die mit Imfinzi behandelt wurden, verglichen mit den Patienten, die Placebo erhielten, waren allgemein über alle analysierten vordefinierten Subgruppen hinweg konsistent.
Fortgeschrittenes kleinzelliges Lungenkarzinom (ES-SCLC) – CASPIAN Studie
Die Wirksamkeit von Imfinzi in Kombination mit Etoposid und entweder Carboplatin oder Cisplatin in therapienaiven ES-SCLC Patienten wurde in CASPIAN, einer randomisierten, offenen, aktiv-kontrollierten, multizentrischen Studie untersucht. Teilnahmeberechtigte Patienten hatten einem Allgemeinzustand nach WHO/ECOG von 0 oder 1, ein Körpergewicht von > 30 kg, waren für den Erhalt einer platinbasierten Chemotherapie als SCLC-Erstlinientherapie geeignet und hatten eine Lebenserwartung von ≥12 Wochen. Auch Patienten mit asymptomatischen oder behandelten Hirnmetastasen, Patienten mit mindestens einer Zielläsion nach RECIST 1.1 sowie einer ausreichenden Organ- und Knochenmarkfunktion waren teilnahmeberechtigt. Die Wahl des Platin-Wirkstoffs lag unter Berücksichtigung der berechneten Kreatinin-Clearance im Ermessen des Prüfarztes. Von einer Teilnahme ausgeschlossen waren Patienten mit anamnestisch bekannter Thoraxbestrahlung oder einer geplanten Konsolidierungs-Thoraxbestrahlung; einem grösseren chirurgischen Eingriff innerhalb von 28 Tagen vor der ersten Dosis; unkontrollierter zwischenzeitlicher Erkrankung; leptomeningealer Karzinomatose; anamnestisch bekannter aktiver primärer Immundefizienz; Autoimmunerkrankungen einschliesslich des paraneoplastischen Syndroms (PNS); aktiver oder dokumentierter Autoimmunkrankheit oder entzündlicher Erkrankungen; Anwendung systemischer Immunsuppressiva innerhalb von 14 Tagen vor der erstmaligen Gabe der Therapie, physiologische Dosen systemischer Kortikosteroide ausgenommen; aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion sowie Patienten, die innerhalb von 30 Tagen vor oder nach Beginn der Behandlung mit Imfinzi einen attenuierten Lebendimpfstoff erhalten hatten bzw. sollten.
Die Randomisierung erfolgte stratifiziert nach der geplanten platinbasierten Therapie in Zyklus 1 (Carboplatin oder Cisplatin).
Die Auswertung der Wirksamkeit bei ES-SCLC stützte sich auf einen Vergleich zwischen den Gruppen:
·Imfinzi 1500 mg und je nach Wahl des Prüfarztes entweder Carboplatin (AUC 5 oder 6 mg/ml/min) oder Cisplatin (75-80 mg/m2) an Tag 1 und Etoposid (80-100 mg/m2) intravenös an den Tagen 1, 2 und 3 jedes 21-tägigen Zyklus über 4 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen, bis zur Krankheitsprogression oder inakzeptablen Toxizität
·Je nach Wahl des Prüfarztes entweder Carboplatin (AUC 5 oder 6 mg/ml/min) oder Cisplatin (75-80 mg/m2) an Tag 1 und Etoposid (80-100 mg/m2) intravenös an den Tagen 1, 2 und 3 jedes 21tägigen Zyklus über bis zu 6 Zyklen. Nach Abschluss der Chemotherapie prophylaktische Schädelbestrahlung (Prophylactic Cranial Irradiation, PCI), nach Ermessen des Prüfarztes
Die Gabe von Imfinzi als Monotherapie war auch nach einer Progression der Erkrankung gestattet, sofern der Patient klinisch stabil und nach Meinung des Prüfers ein klinischer Nutzen zu erwarten war.
Tumorbeurteilungen fanden 6 und 12 Wochen nach der Randomisierung und anschliessend alle 8 Wochen statt, bis eine gesicherte objektive Krankheitsprogression festgestellt wurde. Der Überlebensstatus wurde nach Beendigung der Behandlung 2 monatlich überprüft.
Die primären Endpunkte der Studie waren das Gesamtüberleben (OS) unter Imfinzi + Chemotherapie vs. Chemotherapie allein. Wichtigster sekundärer Endpunkt war das progressionsfreie Überleben (PFS). Weitere sekundäre Endpunkte waren die Objektive Ansprechrate (ORR), OS- und PFS-Meilensteinen sowie Patient Reported Outcomes (PRO). Die Beurteilung des PFS und der ORR erfolgte durch den Prüfarzt gemäss RECIST v1.1.
Eine geplante (primäre) Interimsanalyse zeigte, dass Imfinzi + Chemotherapie vs. Chemotherapie den primären OS-Endpunkt erreichte. Die Ergebnisse sind untenstehend zusammengefasst.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren ausgewogen in beiden Gruppen (268 Patienten in der Imfinzi + Chemotherapie-Gruppe und 269 Patienten in der Chemotherapie-Gruppe). Zu Studienbeginn wies die Gesamtpopulation der Studie folgende Eigenschaften auf: männlich (69,6 %), Alter ≥65 Jahre (39,6 %), medianes Alter 63 Jahre (Bereich: 28 bis 82 Jahre), weiss (83,8 %), asiatisch (14,5 %), schwarz bzw. afroamerikanisch (0,9 %), andere (0,6 %), nicht-hispanisch oder -lateinamerikanisch (96,1 %), aktueller oder ehemaliger Raucher (93,1 %), Nie-Raucher (6,9 %), WHO/ECOG-PS 0 (35,2 %), WHO/ECOG-PS 1 (64,8 %), Stadium IV 90,3 %, 24,6 % der Patienten erhielten Cisplatin und 74,1 % der Patienten erhielten Carboplatin. In der Chemotherapie-Gruppe erhielten 56,8 % der Patienten 6 Zyklen Chemotherapie und 7,8 % der Patienten erhielten PCI.
In der primären Analyse der Studie zeigte sich eine statistisch signifikante Verbesserung des OS unter Imfinzi + Chemotherapie vs. Chemotherapie allein [HR=0,73 (95 %-KI: 0.591, 0.909), p = 0,0047]. Das mediane Gesamtüberleben betrug in der Imfinzi + Chemotherapie-Gruppe 13,0 Monate (95 %-KI: 11.5, 14.8) und in der Chemotherapie-Gruppe10,3 Monate (95 %-KI: 9.3, 11.2). 336 Patienten (155 (57,8 %) in der Imfinzi + Chemotherapie-Gruppe und 181 (67,3 %) in der Chemotherapie-Gruppe) waren verstorben. Das Gesamtüberleben nach 12 resp. 18 Monaten betrug 53,7 % resp. 33,9 % in der Imfinzi + Chemotherapie-Gruppe und 39,8 % resp. 24,7 % in der Chemotherapie-Gruppe.
In der Analyse der Langzeitnachbeobachtung mit einer medianen Nachbeobachtungszeit von 39,3 Monaten zeigte sich unter Imfinzi + Chemotherapie vs. Chemotherapie allein weiterhin eine anhaltende Verbesserung des OS [HR=0,71 (95%-KI: 0,595; 0,858). Das mediane OS betrug in der Imfinzi + Chemotherapie-Gruppe 12,9 Monate (95%-KI: 11,3; 14,7) und 10,5 Monate (95%-KI: 9,3; 11,2) in der Chemotherapie-Gruppe. 469 Patienten (221 (82,5 %) in der Imfinzi + Chemotherapie-Gruppe und 248 (92,2 %) unter Chemotherapie allein) waren verstorben. Das OS nach 24 bzw. 36 Monaten betrug 22,9 % bzw. 17,6 % in der Imfinzi + Chemotherapie-Gruppe und 13,9 % bzw. 5,8 % in der Chemotherapie-Gruppe.
Unter Imfinzi + Chemotherapie war eine Verbesserung des PFS vs. Chemotherapie allein festzustellen [HR=0,78 (95%-KI: 0.645, 0.936) nominaler p-Wert = 0,0078]. 459 Patienten (226 (84,3 %) in der Imfinzi + Chemotherapie-Gruppe und 233 (86,6 %) in der Chemotherapie-Gruppe) waren progredient oder verstorben. Das mediane PFS war 5,1 Monate in der Imfinzi + Chemotherapie-Gruppe und 5,4 Monate in der Chemotherapie-Gruppe. Das PFS nach 6 Monaten betrug 45,4 % in der Imfinzi + Chemotherapie-Gruppe und 45,6 % in Chemotherapie-Gruppe. Das PFS nach 12 Monaten betrug 17,5 % in der Imfinzi + Chemotherapie-Gruppe und 4,7 % in der Chemotherapie-Gruppe.
Das ORR betrug 67,9 % (182/268) in der Imfinzi + Chemotherapie-Gruppe und 57,6 % (155/269) in der Chemotherapie-Gruppe. Es gab 6 Fälle mit komplettem Ansprechen in der Imfinzi + Chemotherapie-Gruppe und 2 Fälle in der Chemotherapie-Gruppe.
Subgruppenanalyse
Die Verbesserung des OS bei Patienten, die Imfinzi + Chemotherapie erhielten, gegenüber den mit Chemotherapie allein behandelten Patienten war konsistent über die verschiedenen nach Demographie, geographischer Region, Carboplatin- oder Cisplatin-Anwendung und Erkrankungsmerkmalen präspezifizierten Subgruppen hinweg zu beobachten.
Explorative Subgruppenanalyse entsprechend der PD-L1-Expression
Die limitierten explorativen Analysen der PD-L1-Expression wurden retrospektiv im Tumorgewebe mit dem VENTANA PD-L1 (SP263) IHC-Test durchgeführt und umfassten Daten sowohl von Immun- (IC) als auch von Tumorzellen (TC) (52,3% des gesamten Analysesets). Der PD-L1-Status der TC wurde durch Membranfärbung gegenüber Hintergrund bestimmt, während der PD-L1-Status der IC durch Färbung gegenüber Hintergrund von tumorassoziierter IC bestimmt wurde. Die Prävalenz der PD-L1-Expression ist gering, wobei eine PD-L1 positive Expression (≥1%) nur bei 25,8% der IC und bei 5,7% der TC beobachtet wurde.
In der IC-Subgruppe zeigten die Resultate der finalen Analyse eine OS HR von 0,64 (95% CI: 0,473, 0,855) bei PD-L1-negativen Patienten (< 1% IC) und eine OS HR von 0,59 (95% CI: 0,342, 1,019) bei PD-L1-positiven Patienten (≥1% IC). In der TC-Subgruppe zeigten die Resultate der finalen Analyse eine OS HR von 0,62 (95% KI: 0,475, 0,810) bei PD-L1-negativen Patienten (< 1% TC) und eine OS HR von 0,75 (95% KI: 0,238, 2,269) bei PD-L1-positiven Patienten (≥1% TC). Insgesamt zeigen diese Daten keinen klaren Zusammenhang zwischen dem PD-L1-Status und der Wirksamkeit in Bezug auf OS.
Biliäres Karzinom – TOPAZ-1-Studie
In der TOPAZ-1-Studie wurde die Wirksamkeit von Imfinzi in Kombination mit Gemcitabin und Cisplatin untersucht. Die TOPAZ-1-Studie war eine multizentrische, randomisierte, placebokontrollierte Doppelblind-Studie mit 685 Patienten mit histologisch gesichertem lokal fortgeschrittenen oder metastasierten biliären Karzinom und einem ECOG-Performance-Status von 0 oder 1. Die Studie schloss auch Patienten ein, die mehr als 6 Monate nach einem chirurgischen Eingriff und/oder Abschluss einer adjuvanten Therapie ein Rezidiv ausgebildet hatten. Die Patienten mussten mindestens eine Zielläsion gemäss RECIST V1.1 und eine adäquate Organ- und Knochenmarkfunktion aufweisen.
Ausgeschlossen waren Patienten mit Karzinom der Ampulla vateri (Papillenkarzinom), aktiver oder zuvor dokumentierter Autoimmunerkrankung oder entzündlicher Erkrankung, HIV-Infektion oder anderer aktiver Infektion (einschliesslich Tuberkulose und Hepatitis C) und solche, die aktuell oder innerhalb von 14 Tagen vor der ersten Dosis von Imfinzi eine immunsuppressive Medikation erhielten.
Die Randomisierung erfolgte stratifiziert nach Krankheitsstatus und Lokalisation des Primärtumors.
Die Patienten wurden im Verhältnis 1:1 randomisiert und den folgenden Behandlungen zugeteilt:
·Arm 1: Imfinzi 1500 mg intravenös an Tag 1 + Gemcitabin 1000 mg/m2 und Cisplatin 25 mg/m2 (jeweils verabreicht an den Tagen 1 und 8) alle 3 Wochen (21 Tage) für bis zu 8 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen, solange ein klinischer Nutzen zu beobachten ist oder bis eine inakzeptable Toxizität auftritt, oder
·Arm 2: Placebo intravenös an Tag 1 + Gemcitabin 1000 mg/m2 und Cisplatin 25 mg/m2 (jeweils verabreicht an den Tagen 1 und 8) alle 3 Wochen (21 Tage) für bis zu 8 Zyklen, gefolgt von Placebo alle 4 Wochen, solange ein klinischer Nutzen zu beobachten ist oder bis eine inakzeptable Toxizität auftritt.
Tumorbeurteilungen erfolgten in den ersten 24 Wochen nach dem Tag der Randomisierung alle 6 Wochen und dann alle 8 Wochen bis zu einer bestätigten objektiven Krankheitsprogression.
Der primäre Endpunkt der Studie war OS und der wichtigste sekundäre Endpunkt PFS. Weitere sekundäre Endpunkte waren ORR, Ansprechdauer (DoR) und Daten aus Patientenfragebogen (Patient Reported Outcomes, PRO). Die Beurteilung von PFS, ORR und DoR erfolgte durch den Prüfarzt gemäss RECIST V1.1.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren in den beiden Studienarmen ausgewogen (341 Patienten in Arm 1 und 344 Patienten in Arm 2). Zu Studienbeginn wies die Gesamtpopulation der Studie folgende demographische Merkmale auf: männlich (50,4 %), Alter < 65 Jahre (53,3 %), weiss (37,2 %), asiatisch (56,4 %), schwarz oder afroamerikanisch (2,0 %), andere (4,2 %), nichthispanisch oder -lateinamerikanisch (93,1 %), ECOG PS 0 (49,1 %) vs. PS 1 (50,9 %), Lokalisation des Primärtumors intrahepatisches Cholangiokarzinom (55,9 %), extrahepatisches Cholangiokarzinom (19,1 %) und Gallenblasenkarzinom (25,0 %), Krankheitsstatus rezidiviert (19,1 %) vs. initial nicht resezierbar (80,7 %), metastasiert (86,0 %) vs. lokal fortgeschritten (13,9 %).
Die Studie zeigte eine statistisch signifikante Verbesserung des OS und des PFS bei einer vorab geplanten Zwischenanalyse basierend auf einer Lan-DeMets-Alpha-Spending-Funktion mit O'Brien-Fleming-Grenze und der tatsächlichen Anzahl an beobachteten Ereignissen (Lan und DeMets, 1983) [HR = 0,80 (95%-KI: 0,66, 0,97), p=0,021 für das OS und HR=0,75 (95%-KI: 0,63, 0,89), p=0,001 für das PFS]. Die Reife in Bezug auf das OS betrug 61,9 %, und die Reife in Bezug auf das PFS betrug 83,6 %. Die Ergebnisse dieser Analyse für das PFS sind in Tabelle 5 aufgeführt.
Eine weitere OS-Analyse wurde 6,5 Monate nach der Zwischenanalyse mit einer OS-Reife von 76,9 % durchgeführt. Die HR für das OS betrug 0,76 (95%-KI: 0,64, 0,91) und das mediane Überleben betrug 12,9 Monate (95%-KI: 11,6, 14,1). Die Ergebnisse dieser OS-Analyse sind in Tabelle 5 aufgeführt.
Tabelle 5. Wirksamkeitsergebnisse der TOPAZ-1-Studie
|
|
Imfinzi + Gemcitabin und Cisplatin
(n = 341)
|
Placebo + Gemcitabin und Cisplatin
(n = 344)
| |
OS (DCO: 25 Feb 2022)
|
| |
Anzahl der Todesfälle (%)
|
248 (72,7)
|
279 (81,1)
| |
Medianes OS (Monate) (95%-KI)a
|
12, 9
(11,6; 14,1)
|
11, 3
(10,1; 12,5)
| |
HR (95%-KI)b
|
0, 76 (0,64; 0,91)
| |
OS nach 24 Monaten (%) (95%-KI)a
|
23,6(18,7; 28,9)
|
11,5(7,6; 16,2)
| |
PFS (DCO: 11 Aug 2021)
|
| |
Anzahl der Ereignisse (%)
|
276 (80,9)
|
297 (86,3)
| |
Medianes PFS (Monate)
(95%-KI)a
|
7,2
(6,7; 7,4)
|
5,7
(5,6; 6,7)
| |
HR (95%-KI)b
|
0,75 (0,63; 0,89)
| |
p-Wertb,c
|
0,001
|
a Mit der Kaplan-Meier-Methode berechnet. KI für den Median mit der Brookmeyer-Crowley-Methode generiert.
b Die Analyse für HR erfolgte mit einem stratifizierten Cox-Proportional-Hazards-Modell und der 2seitige p-Wert basiert auf einem stratifizierten Log-Rank-Test; beide sind für Krankheitsstatus und Lokalisation des Primärtumors adjustiert.
c p-Wert basierend auf den Ergebnissen der vorab geplanten Zwischenanalyse. Auf der Basis einer Alpha-Spending-Funktion nach Lan-DeMets mit Pocock-Grenze und der tatsächlich beobachteten Anzahl an Ereignissen.
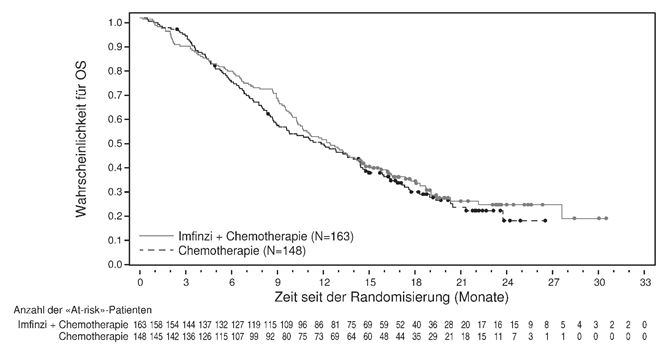
Es wurde eine Subgruppenanalyse nach geographischer Region durchgeführt, die sich auf Patienten stützte, die in Prüfzentren in Asien randomisiert wurden (178 Patienten für Durvalumab + Gem/Cis und 196 Patienten für Placebo + Gem/Cis), sowie auf Patienten, die in weltweit verteilten Prüfzentren randomisiert wurden (163 Patienten bzw. 148 Patienten). Die Hazard Ratio für das OS (95% KI) betrug 0,68 (0,54, 0,85) in Asien und 0,91 (95% KI: 0,70, 1,18) im Rest der Welt (siehe Abbildungen 1 und 2).
Abbildung 1: Kaplan-Meier Kurve für das OS in Asien (DCO: 25 Feb 2022)
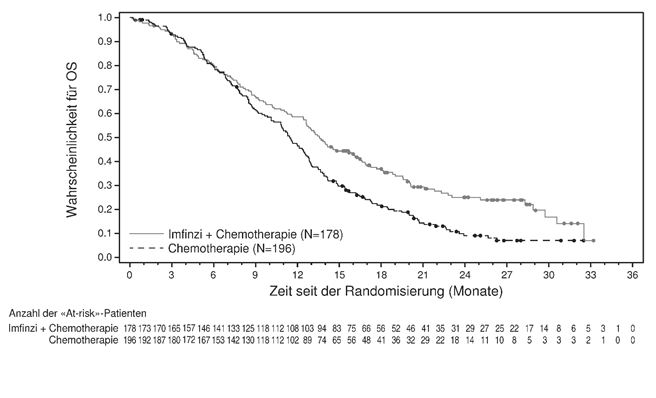
Abbildung 2: Kaplan-Meier-Kurve für das OS im Rest der Welt (DCO: 25 Feb 2022)
Muskelinvasiver Blasenkrebs (MIBC) – NIAGARA-Studie
NIAGARA war eine randomisierte, offene, multizentrische Phase-III-Studie zur Untersuchung der Wirksamkeit von neoadjuvantem Imfinzi in Kombination mit Gemcitabin und Cisplatin, gefolgt von adjuvanter Imfinzi-Monotherapie bei Patienten mit MIBC. In der Studie wurden 1063 für Cisplatin geeignete Patienten mit klinischem Tumorstadium T2-T4aN0/1M0 randomisiert, die für eine radikale Zystektomie in Frage kamen und zuvor keine systemische Chemotherapie oder immunvermittelte Therapie zur Behandlung von MIBC erhalten hatten. Es konnten Patienten mit angemessener (Kreatinin-Clearance [CrCl] ≥60 ml/min) oder grenzwertiger Nierenfunktion (CrCl ≥40 ml/min bis < 60 ml/min) teilnehmen. Von der Studie ausgeschlossen waren Patienten mit ausschliesslich nichturothelialer Histologie, mit jedweder kleinzelligen Histologie und mit primärem Nicht-Blasen-Krebs (d.h. Harnleiter, Harnröhre oder Nierenbecken) des Urothels, mit aktiver oder früher dokumentierter Autoimmunerkrankung, aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion oder Einnahme von immunsuppressiven Medikamenten innerhalb von 14 Tagen vor der ersten Dosis von Durvalumab mit Ausnahme von systemischen Kortikosteroiden, wenn in physiologischen Dosen oder als Prämedikation verwendet.
Die Randomisierung erfolgte stratifiziert nach klinischem Tumorstadium T2N0 vs. > T2N0 (einschliesslich T2N1, T3 und T4a), Nierenfunktion (angemessene Nierenfunktion: CrCl ≥60 mlL/min vs. grenzwertige Nierenfunktion: CrCl ≥40 ml/min und < 60 ml/min), und PD-L1-Expressionsstatus (hoch vs. niedrig/negativ).
Die Patienten wurden im Verhältnis 1:1 auf perioperatives Imfinzi mit neoadjuvanter Chemotherapie (Arm 1) oder neoadjuvante Chemotherapie allein (Arm 2) randomisiert.
·Arm 1 (Imfinzi + Chemotherapie): Imfinzi 1500 mg + Gemcitabin 1000 mg/m² und Cisplatin 70 mg/m² alle 3 Wochen für 4 Zyklen vor der Operation, gefolgt von Imfinzi 1500 mg alle 4 Wochen für bis zu 8 Zyklen nach der Operation, oder
·Arm 2 (Chemotherapie): Gemcitabin 1000 mg/m² und Cisplatin 70 mg/m² alle 3 Wochen für 4 Zyklen vor der Operation, ohne postoperative Behandlung.
Patienten mit grenzwertiger Nierenfunktion erhielten eine geteilte Dosis Cisplatin von 35 mg/m² an den Tagen 1 und 8 eines jeden Zyklus.
Eine RECIST-1.1-Tumorbeurteilung wurde zu Studienbeginn und nach Abschluss der neoadjuvanten Therapie (vor der Operation) durchgeführt. Nach der Operation wurden in den ersten 24 Monaten alle 12 Wochen, dann 36 Monate lang alle 24 Wochen und danach bis zur Krankheitsprogression, zum Ende der Studie oder zum Tod alle 52 Wochen RECIST-1.1-Tumorbeurteilungen durchgeführt.
Die primären Endpunkte waren pathologisch vollständiges Ansprechen (pathological complete response; pCR) durch verblindete zentrale pathologische Überprüfung und ereignisfreies Überleben (event-free survival; EFS), was eine Beurteilung durch verblindete unabhängige zentrale Überprüfung (BICR) einschloss. Der wichtigste sekundäre Endpunkt war das Gesamtüberleben (overall survival; OS).
Die demografischen Daten und die Merkmale der Erkrankung zu Studienbeginn waren zwischen den 533 Patienten in Arm 1 und den 530 Patienten in Arm 2 grundsätzlich gut ausgeglichen. Die demografischen Daten zu Studienbeginn waren wie folgt: männlich (81,8 %), Alter < 65 Jahre (46,9 %), weiss (67 %), asiatisch (27,9 %), hispanisch oder lateinamerikanisch (8,0 %), schwarz oder afroamerikanisch (0,9 %), andere (0,8 %), und ECOG PS 0 (78,4 %) vs. PS 1 (21,6 %). Die Merkmale der Erkrankung waren wie folgt: Tumorstadium T2N0 (40,3 %) und > T2N0a (59,7 %), regionale Lymphknoten N0 (94,5 %) und N1 (5,5 %), angemessene Nierenfunktion (81,1 %) und grenzwertige Nierenfunktion (18,9 %) und PD-L1-Expressionsstatus hoch (73,1 %) und niedrig/negativ (26,9 %). Zu den histologischen Subtypen gehörten Urothelkarzinom (84,5 %), Urothelkarzinom mit Plattenepithel-Differenzierung (8,2 %), Urothelkarzinom mit varianter Histologie (5,0 %) und Urothelkarzinom mit glandulärer Differenzierung (2,4 %).
In der zweiten präspezifizierten Zwischenanalyse zeigte die Studie eine statistisch signifikante Verbesserung des Überlebens im Imfinzi+Chemotherapie-Arm im Vergleich zum Chemotherapie-Arm [HR = 0,68 (95%-KI: 0,56; 0,82), p < 0,0001]. Die Studie zeigte auch eine statistisch signifikante Verbesserung des OS im Imfinzi+Chemotherapie-Arm im Vergleich zum Chemotherapie-Arm [HR = 0,75 (95%-KI: 0,59; 0.93), p = 0,0106]. Eine numerische Verbesserungder pCR-Raten ,welche jedoch nicht statistisch signifikant war, wurde im Imfinzi + Chemotherapie-Arm [Ansprechrate = 37,3 % (95%-KI: 33,2; 41,6)] im Vergleich zum Chemotherapie-Arm [Ansprechrate = 27,5 % (95%-KI: 23.8, 31.6)] beobachtet. Siehe Tabelle 6.
Tabelle 6: Wirksamkeitsergebnisse der NIAGARA-Studie
|
|
Imfinzi + Chemotherapie
(n = 533)
|
Chemotherapie
(n = 530)
| |
EFSa
| |
Anzahl der Ereignisse (%)
|
187 (35,1)
|
246 (46,4)
| |
Medianes EFS (Monate) (95% KI)b
|
NR (NR; NR)
|
46,1 (32,2; NR)
| |
HR (95% KI)c
|
0,68 (0,56; 0,82)
| |
2-seitiger p-Wertd,e
|
<0,0001
| |
EFS nach 24 Monaten (%) (95% KI)b
|
67,8 (63,6; 71,7)
|
59,8 (55,4; 64,0)
| |
pCRf
| |
Anzahl der Patienten mit Ansprechen
|
199
|
146
| |
Ansprechrate, % (95% KI)g
|
37,3 (33,2; 41,6)
|
27,5 (23,8; 31,6)
| |
Odds Ratio (95% KI)h
|
1,60 (1,23; 2,09)
| |
OSa
| |
Anzahl Ereignisse (%)
|
136 (25,5)
|
169 (31,9)
| |
Medianes OS (Monate) (95% KI)b
|
NR (NR; NR)
|
NR (NR; NR)
| |
HR (95% KI)c
|
0,75 (0,59; 0,93)
| |
2seitiger p-Wertd,e
|
0,0106
| |
OS nach 24 Monaten (%) (95% KI)b
|
82,2 (78,7; 85,2)
|
75,2 (71,3; 78.8)
|
a Die Ergebnisse basieren auf einer vordefinierten Zwischenanalyse (DCO: 29. April 2024), die 68 Monate nach Beginn der Studie durchgeführt wurde.
b Berechnet mittels der Kaplan-Meier-Methode.
c Basierend auf einem stratifizierten Cox-Proportional-Hazard-Modell mit Tumorstadium [T2N0 vs. > T2N0], Nierenfunktion [angemessen vs. grenzwertig] und PD-L1-Status [hoch vs. niedrig/negativ] als Stratifikationsfaktoren.
d Basierend auf dem stratifizierten Log-Rank-Test mit Tumorstadium [T2N0 vs. > T2N0], Nierenfunktion [angemessen vs. grenzwertig] und PD-L1-Status [hoch vs. niedrig/negativ] als Stratifikationsfaktoren.
e Die Grenze für die Erklärung der statistischen Signifikanz für die primären Wirksamkeitsendpunkte pCR-Rate, EFS und den wichtigsten sekundären Endpunkt OS wurde durch ein multiples Testverfahren mit einer alpha-exhaustiven Recycling-Strategie bestimmt. Das Alpha, das bei der Zwischenanalyse dem EFS und OS zugeordnet wurde, basierte auf einer Lan-DeMets Alpha-Ausgabefunktion mit O'Brien-Fleming-Ansatz. (pCR = 0,001, EFS = 0,0412, OS = 0,0154, 2-seitig).
f Basierend auf einer aktualisierten, deskriptiven Analyse des primären Endpunkts. Bei der früheren vorspezifizierten abschliessenden Analyse der pCR (DCO: 14. Jan. 2022) wurde eine numerische Verbesserung der pCR-Raten in dem Imfinzi+Chemotherapie-Arm [Ansprechrate = 33,8 % (95%-KI: 29,8; 38,0)] im Vergleich zum Chemotherapie-Arm [Ansprechrate = 25,8 % (95%-KI: 22,2; 29,8)] [Odds Ratio 1,49 (95%-KI: 1,14; 1,96), p = 0,0038] beobachtet.
g Das KI wurde mit der Clopper-Pearson-Methode berechnet.
h Ermittelt durch logistische Regression, bereinigt um die Stratifikationsfaktoren (Nierenfunktion [angemessen vs. grenzwertig], Tumorstadium [T2N0 vs. > T2N0] und PD-L1-Status [hoch vs. niedrig/negativ] per IVRS).
KI = Konfidenzintervall, HR = Hazard Ratio, NR = Nicht erreicht
|