ZusammensetzungWirkstoffe
Burosumab (aus gentechnologisch veränderten CHO (chinese hamster ovary)-Zellen hergestellt)
Hilfsstoffe
Lhistidinum, sorbitolum (E420) 45.91 mg, polysorbatum 80, Lmethioninum, acidum hydrochloridum, aqua ad iniectabile q.s. ad solutionem pro 1 ml.
Indikationen/AnwendungsmöglichkeitenCRYSVITA ist indiziert für die Behandlung von X-chromosomaler Hypophosphatämie (XLH) bei Erwachsenen, Jugendlichen und Kindern ab 1 Jahr.
Dosierung/AnwendungDie Behandlung sollte von einem Arzt mit Erfahrung bei der Behandlung metabolischer Knochenerkrankungen initiiert und überwacht werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Die Behandlung soll nur dann initiiert werden, wenn die Nüchternkonzentration des Serumphosphats unter dem altersbezogenen Referenzbereich liegt.
Orale Phosphatpräparate und aktive Vitamin D-Derivate (z.B. Calcitriol) müssen 1 Woche vor Behandlungsbeginn abgesetzt werden. Im Verlauf der Therapie kann eine Vitamin D-Substitution mit inaktiven Vorläufern erwogen werden, um dem erhöhten Bedarf infolge der hohen Vitamin D-Aktivierungsrate unter Behandlung mit Burosumab abzudecken. Um eine Hypervitaminose zu vermeiden, sollte der Vitamin D-Spiegel regelmässig kontrolliert werden.
Art der Anwendung
Zur subkutanen Anwendung.
Burosumab soll in Oberarm, Bauch, Gesäss oder Oberschenkel injiziert werden.
Vor der Verabreichung sollte CRYSVITA aus dem Kühlschrank genommen und auf Raumtemperatur gebracht werden.
Die Höchstmenge des Arzneimittels pro Injektionsstelle beträgt 1.5 ml. Wenn an einem Behandlungstag eine grössere Menge als 1.5 ml gegeben werden muss, muss die Gesamtmenge des Arzneimittels aufgeteilt und an zwei oder mehreren verschiedenen Injektionsstellen injiziert werden. Die Injektionsstellen sollen immer wieder gewechselt und sorgfältig auf Anzeichen für mögliche Reaktionen überwacht werden.
Die Injektionen können bis zu 3 Tage vor oder nach dem vorgesehenen Behandlungsdatum verabreicht werden. Innerhalb dieses Intervalls hat eine Änderung des Injektionstages keinen Einfluss auf das Dosierungsschema, d.h. die nächste Injektion kann am ursprünglich vorgesehenen Tag (unter Beibehaltung des entsprechenden Wochentages) durchgeführt werden (siehe auch "versäumte Anwendung" unten).
Dosierung bei Jugendlichen und Kindern ab 1 Jahr
Die Dosis von Burosumab wird titriert mit dem Ziel, Nüchtern-Serumphosphatwerte innerhalb des altersbezogenen Referenzbereichs zu erreichen. Um das Risiko einer ektopen Mineralisation zu minimieren, sollten nach Möglichkeit Nüchtern-Serumphosphatwerte am unteren Ende des Referenzbereichs angestrebt werden.
Therapieeinleitung
Die empfohlene Anfangsdosis bei Kindern und Jugendlichen beträgt 0.4 mg/kg Körpergewicht, alle zwei Wochen verabreicht.
Kinder mit einem Körpergewicht bis zu 15 kg können die Behandlung mit einer Dosis von 5 mg beginnen. Bei Kindern mit einem Körpergewicht ≥15 kg sollte die Dosis auf die nächsten 10 mg gerundet werden. Die Mindestanfangsdosis beträgt 5 mg, die Höchstdosis 90 mg.
Erhaltungstherapie
Der Nüchtern-Serumphosphatwert sollte im ersten Behandlungsmonat alle 2 Wochen bestimmt werden, danach für die nächsten 3 Monate alle 4 Wochen, anschliessend nach Bedarf. Wenn der Serumphosphatwert innerhalb des altersbezogenen Referenzbereichs liegt, soll die gewählte Dosis beibehalten werden. Um den Serumphosphatwert innerhalb des unteren altersbezogenen Referenzbereichs zu halten, ist das nachfolgende Schema für die Dosisanpassung zu befolgen.
Dosiserhöhung
Liegt der Nüchtern-Serumphosphatwert 4 Wochen nach der Anfangsdosis oder im weiteren Verlauf unterhalb des altersbezogenen Referenzbereichs, kann die Dosis schrittweise allmählich bis zu einer Höchstdosis von 2.0 mg/kg Körpergewicht (maximal 90 mg) alle 2 Wochen erhöht werden. Bei Kindern mit einem Körpergewicht von ≥15 kg sollen alle Dosen auf die nächsten 10 mg gerundet werden. Der Nüchtern-Serumphosphatspiegel soll 4 Wochen nach jeder Dosisanpassung kontrolliert werden. Die Burosumab-Dosierung soll nicht häufiger als einmal alle 4 Wochen angepasst werden. Die nachfolgende Tabelle 1 zeigt mögliche initiale Dosiserhöhungs-Schritte auf.
Tabelle 1: Dosierungsschema für eine schrittweise Dosiserhöhung bei Jugendlichen und Kindern ab 1 Jahr
|
Körpergewicht (kg)
|
Anfangsdosis (mg)
(0.4 mg/kg)
|
Erste Dosiserhöhung auf (mg)
(0.8 mg/kg)
|
Zweite Dosiserhöhung auf (mg)
(1.2 mg/kg)*
| |
< 15
|
5
|
10
|
15
| |
15 – 18
|
10
|
15
|
20
| |
19 – 31
|
10
|
20
|
30
| |
32 – 43
|
20
|
30
|
40
| |
44 – 56
|
20
|
40
|
60
| |
57 – 68
|
30
|
50
|
70
| |
69 – 80
|
30
|
60
|
90
| |
81 – 93
|
40
|
70
|
90
| |
94 – 105
|
40
|
80
|
90
| |
106 und mehr
|
40
|
90
|
90
|
* Die Tabelle zeigt die Dosiserhöhung bis 1.2 mg/kg auf. Weitere Dosiserhöhungen auf maximal 2.0 mg/kg bzw. 90 mg Höchstdosis sollen vom verschreibenden Arzt berechnet werden, wobei der geringere Wert massgeblich ist.
Dosisunterbrechung/reduktion
Liegt der Nüchtern-Serumphosphatwert 2 Wochen nach der Anfangsdosis oder im weiteren Behandlungsverlauf über dem altersbezogenen Referenzbereich, soll die nächste Dosis ausgesetzt und der Nüchtern-Serumphosphatwert innerhalb von 4 Wochen erneut bestimmt werden. Die Behandlung mit Burosomab darf erst dann wieder aufgenommen werden, wenn der Nüchtern-Serumphosphatwert unterhalb des altersbezogenen Referenzbereich liegt. Sobald der Serumphosphatwert unter dem altersbezogenen Referenzbereich liegt, kann die Behandlung mit der Hälfte der vorherigen Dosis bis zu einer maximalen (Gesamt-)Dosis von 40 mg, – alle zwei Wochen verabreicht – wiederaufgenommen werden. Der Nüchtern-Serumphosphatwert ist 4 Wochen nach der Dosisanpassung erneut zu bestimmen und die Dosisanpassung gemäss Tabelle 2 vorzunehmen.
Tabelle 2: Dosierungsschema für die Wiederaufnahme der Therapie nach einer Unterbrechung wegen Überschreitung des Phosphat-Normbereiches bei Jugendlichen und Kindern ab 1 Jahr
|
Vorherige Dosis (mg)
|
Dosis bei Wiederaufnahme (mg)
| |
10
|
5
| |
15
|
10
| |
20
|
10
| |
30
|
10
| |
40
|
20
| |
50
|
20
| |
60
|
30
| |
70
|
30
| |
80
|
40
| |
90
|
40
|
Übergang auf die Erwachsenen-Dosierung
Bei Vollendung des 18. Lebensjahrs sollte die Dosierung auf das nachfolgend beschriebene Dosierungsschema für Erwachsene umgestellt werden.
Dosierung bei Erwachsenen
Therapieeinleitung
Die empfohlene Anfangsdosis bei Erwachsenen beträgt 1 mg/kg Körpergewicht alle 4 Wochen, gerundet auf die nächsten 10 mg, bis zu einer Höchstdosis von 90 mg.
Erhaltungstherapie
2 Wochen nach der ersten Burosumab-Injektion soll der Nüchtern-Serumphosphatwert bestimmt werden, dann für die nächsten 3 Monate alle 4 Wochen, anschliessend nach Bedarf. Wenn der Nüchtern-Serumphosphatwert im angestrebten Zielbereich liegt, soll die gewählte Dosis beibehalten werden.
Dosisanpassung
Liegt der Nüchtern-Serumphosphatwert über dem Normbereich, soll die nächste Dosis ausgesetzt und der Nüchtern-Serumphosphatwert nach 4 Wochen erneut gemessen werden. Die Behandlung mit Burosumab darf erst dann wieder aufgenommen werden, wenn der Nüchtern-Serumphosphatwert unterhalb des Normbereichs liegt. Sobald der Serumphosphatwert unterhalb des Normbereichs liegt, kann die Behandlung mit der Hälfte der ursprünglichen Anfangsdosis (gemäss Tabelle 3) wieder aufgenommen werden bis zu einer Maximaldosis von 40 mg alle 4 Wochen, wobei die Dosen auf die nächsten 10 mg gerundet werden. Der Nüchtern-Serumphosphatwert soll 2 Wochen nach einer Dosisänderung erneut bestimmt werden.
Tabelle 3: Dosierungsschema für die Wiederaufnahme der Therapie nach einer Unterbrechung wegen Überschreitung des Phosphat-Normbereiches bei Erwachsenen
|
Vorherige Dosis (mg)
|
Dosis bei Wiederaufnahme (mg)
| |
40
|
20
| |
50
|
20
| |
60
|
30
| |
70
|
30
| |
80 und mehr
|
40
|
Versäumte Anwendung
Wird eine Dosis versäumt, sollte die Injektion so bald wie möglich nachgeholt werden. Es konnte gezeigt werden, dass bei einer Abweichung um +/-3 Tage vom vorgesehenen Injektionstermin Wirksamkeit und Sicherheit der Therapie nicht in relevanter Weise beeinflusst werden. Anschliessend kann die Verabreichung entsprechend dem normalen Dosierungsintervall fortgesetzt werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Burosumab wurde bei Patienten mit Leberinsuffizienz nicht spezifisch untersucht. Es sind daher keine Dosierungsempfehlungen möglich.
Patienten mit Nierenfunktionsstörungen
Zu Patienten mit eingeschränkter Nierenfunktion liegen nur begrenzte Daten vor. Burosumab darf bei Patienten mit schwer eingeschränkter Nierenfunktion oder terminaler Niereninsuffizienz nicht angewendet werden (siehe “Kontraindikationen”).
Ältere Patienten
Bei älteren Patienten liegen nur begrenzte Daten vor.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Burosumab bei Kindern unter einem Jahr ist nicht erwiesen.
Kontraindikationen·Gleichzeitige Anwendung mit oralen Phosphatpräparaten und/oder aktiven Vitamin D-Derivaten.
·Nüchtern-Serumphosphatwert zum Zeitpunkt der Behandlungseinleitung innerhalb oder oberhalb des altersbezogenen Normbereichs.
·Schwer eingeschränkte Nierenfunktion oder terminale Niereninsuffizienz.
·Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenEktopische Mineralisierung
Eine ektopische Mineralisierung, die sich in Form einer Nephrokalzinose manifestiert, wurde bei Patienten mit XLH beobachtet, die mit oralen Phosphatpräparaten und aktiven Vitamin D-Derivaten behandelt wurden. Diese Arzneimittel müssen mindestens 1 Woche vor Einleitung der Burosumab-Behandlung abgesetzt werden (siehe “Dosierung/Anwendung”).
Eine Überwachung des Patienten auf Anzeichen und Symptome von Nephrokalzinose, z. B. mittels Nieren-Ultrasonographie, wird zu Behandlungsbeginn sowie während der ersten 12 Behandlungsmonate alle 6 Monate empfohlen. Danach sollen jährliche Kontrollen stattfinden. Es wird eine Überwachung der alkalischen Phosphatase-, Kalzium-, Parathormon (PTH)- und Kreatinin-Werte im Plasma alle 6 Monate (bei Kindern von 1-2 Jahren alle 3 Monate) oder je nach Bedarf empfohlen.
Zu einer Kontrolle des Kalzium- und Phosphat-Wertes im Urin alle 3 Monate wird geraten.
Hyperphosphatämie
Der Nüchtern-Serumphosphatwert des Patienten muss wegen des Risikos einer Hyperphosphatämie überwacht werden. Zur Senkung des Risikos einer ektopischen Mineralisierung wird empfohlen, einen Nüchtern-Serumphosphatwert im unteren Bereich des altersentsprechenden Normbereichs anzustreben. Es kann eine Dosisunterbrechung und/oder reduktion erforderlich sein (siehe “Dosierung/Anwendung”). Eine regelmässige Messung des postprandialen Serumphosphats ist ratsam.
Serumparathormon
Bei manchen Patienten mit XLH wurden während der Burosumab-Behandlung Anstiege des Parathormons im Serum beobachtet. Es wird dazu geraten, den Serumparathormon-Wert in regelmässigen Abständen zu kontrollieren.
Reaktionen an der Injektionsstelle
Die Anwendung von Burosumab kann zu lokalen Reaktionen an der Injektionsstelle führen. Bei jedem Patienten, der schwere Reaktionen an der Injektionsstelle zeigt, sollte die Behandlung unterbrochen werden.
Überempfindlichkeit
Therapeutische Proteine, wie Burosumab, können mit Überempfindlichkeitsreaktionen verbunden sein. In klinischen Studien wurden leichte oder mässige Überempfindlichkeitsreaktionen (z.B. Hautausschlag, Ausschlag an der Injektionsstelle) beobachtet (siehe "Unerwünschte Wirkungen").
Bei Auftreten schwerwiegender Überempfindlichkeitsreaktionen muss Burosumab abgesetzt werden.
Hilfsstoff von besonderem Interesse
Dieses Arzneimittel enthält 45.91 mg Sorbitol pro Durchstechflasche; dies entspricht 45.91 mg/ml.
Die additive Wirkung gleichzeitig angewendeter Sorbitol (oder Fructose) –haltiger Arzneimittel und die Einnahme von Sorbitol (oder Fructose) über die Nahrung ist zu berücksichtigen.
InteraktionenMit Burosumab wurden keine Studien zu pharmakokinetischen Interaktionen durchgeführt.
Schwangerschaft, StillzeitSchwangerschaft
Es gibt keine Daten zur Anwendung von Burosumab bei Schwangeren. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»). Therapeutische Proteine können die Plazenta passieren. Die Anwendung von CRYSVITA während der Schwangerschaft und bei Frauen im gebärfähigen Alter, welche nicht verhüten, wird nicht empfohlen.
Stillzeit
Es ist nicht bekannt, ob Burosumab / Metaboliten in die Muttermilch übergehen. Bisher wurden keine Studien zu den Auswirkungen von Burosumab auf die Milchproduktion oder sein Vorliegen in der Muttermilch durchgeführt. Es ist bekannt, dass menschliches IgG in der Muttermilch vorhanden ist. Ein Risiko für das Neugeborene / Kind kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit CRYSVITA verzichtet werden soll/die Behandlung mit CRYSVITA zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine klinischen Daten über die Auswirkungen von Burosumab auf die Fertilität des Menschen vor. Tierexperimentelle Studien haben eine ektopische Mineralisierung einschliesslich Auswirkungen auf die männlichen Fortpflanzungsorgane gezeigt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt. Nach der Anwendung von Burosumab kann Schwindel auftreten. Aufgrunddessen kann die Fahrtüchtigkeit und das Bedienen von Maschinen beeinträchtigt sein. Die Patienten sind zu entsprechender Vorsicht anzuhalten.
Unerwünschte WirkungenNachfolgend sind die unerwünschten Wirkungen nach Systemorganklasse (gemäss MedDRA) und Häufigkeitskategorien aufgeführt, die in klinischen Studien und/oder nach der Markteinführung unter der Anwendung von Burosumab beobachtet wurden. Die Häufigkeiten sind dabei gemäss folgender Konvention definiert sind: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1000, < 1/100), selten (≥ 1/10'000, < 1/1000), sehr selten (< 1/10’000) und nicht bekannt (basierend überwiegend auf Spontanmeldungen aus der Marktüberwachung, genaue Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Unerwünschte Wirkungen bei pädiatrischen Patienten
Die häufigsten unerwünschten Arzneimittelwirkungen (UAW), die bei pädiatrischen Patienten während bis zu 64 Behandlungswochen in klinischen Studien gemeldet wurden, waren Reaktionen an der Injektionsstelle (56%), Husten (56%), Kopfschmerzen (50%), Fieber (43%) und Schmerzen in einer Extremität (40%).
Tabelle 4: Unerwünschte Wirkungen bei pädiatrischen Patienten (Alter > 1 Jahr)
|
Systemorganklasse
|
Sehr häufig
|
Häufig
|
Nicht bekannt
| |
Infektionen und parasitäre Erkrankungen
|
Zahnabszess1 (35%)
|
|
| |
Stoffwechsel- und Ernährungsstörungen
|
Vitamin D-Mangel2 (32%)
|
|
erhöhte Phosphatwerte im Blut3
| |
Erkrankungen des Nervensystems
|
Kopfschmerzen (50%)
|
Schwindel4
|
| |
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
|
Husten5 (56%)
|
|
| |
Erkrankungen des Gastrointestinaltrakts
|
Erbrechen (39%),
Diarrhoe (25%),
Übelkeit (15%),
Obstipation (11%),
Karies (11%)
|
|
| |
Erkrankungen der Haut und des Unterhautzellgewebes
|
Ausschlag6 (24%)
|
|
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Schmerzen in einer Extremität (40%),
Myalgien (11%)
|
|
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Reaktionen an der Injektionsstelle7 (56%),
Fieber (43%)
|
|
|
1Zahnabszess umfasst: Zahnabszess, Zahninfektion und Zahnschmerzen
2Erniedrigtes Vitamin D umfasst: Vitamin D-Mangel, 25-Hydroxycholecalciferol im Blut erniedrigt und Vitamin D erniedrigt
3Erhöhte Phosphorwerte im Blut umfasst: Erhöhte Phosphorwerte im Blut und Hyperphosphatämie
4Schwindel umfasst: Schwindelgefühl und Schwindelgefühl bei Belastung
5Husten umfasst: Husten und Husten mit Auswurf
6Ausschlag umfasst: Ausschlag, erythematöser Hautausschlag, Ausschlag generalisiert, Ausschlag mit Juckreiz, Ausschlag makulo-papulös und Ausschlag pustulös
7Reaktion an der Injektionsstelle umfasst: Reaktion an der Injektionsstelle, Erythem an der Injektionsstelle, Jucken an der Injektionsstelle, Schwellung an der Injektionsstelle, Schmerzen an der Injektionsstelle, Ausschlag an der Injektionsstelle, blauer Fleck an der Injektionsstelle, Verfärbung an der Injektionsstelle, Beschwerden an der Injektionsstelle, Injektionsstelle Hämatom, Blutung an der Injektionsstelle, Verhärtung an der Injektionsstelle, Makula an der Injektionsstelle und Urtikaria an der Injektionsstelle
Beschreibung spezifischer unerwünschter Wirkungen bei Kindern und Zusatzinformationen
Reaktionen an der Injektionsstelle
Etwa 56% der Patienten zeigten eine Reaktion an der Injektionsstelle. Die Reaktionen an der Injektionsstelle traten meist innerhalb von 1 Tag nach der Injektion des Arzneimittels auf, hielten etwa 1 bis 3 Tage an, erforderten keine Behandlung und klangen in fast allen Fällen wieder ab.
Überempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen (wie Ausschlag an der Injektionsstelle, Ausschlag, Urtikaria, Gesichtsschwellung, Dermatitis) wurden von 18% der Patienten berichtet. Alle berichteten Überempfindlichkeitsreaktionen waren von leichtem bis mittlerem Schweregrad, erforderten keine spezifische Behandlung und traten bei fortgesetzter Behandlung im Allgemeinen nicht erneut auf.
Immunogenität
Bei 6% der mit Burosumab behandelten Patienten wurden gegen das Arzneimittel gerichtete Antikörper (ADA, Anti-Drug Antibodies) nachgewiesen. Diese Patienten waren jedoch bereits vor der Behandlung positiv auf ADA getestet worden. Ein Zusammenhang mit einer Häufung unerwünschter Ereignisse oder einem Wirksamkeitsverlust war nicht erkennbar.
Erniedrigte Vitamin D-Spiegel
Nach Einleitung der Behandlung mit Burosumab wurden bei ca. 8% der Patienten verminderte 25-Hydroxy-Vitamin D-Spiegel (inaktives Vitamin D) im Serum beobachtet, möglicherweise aufgrund einer erhöhten Umwandlung zu aktiviertem Vitamin D. Die Plasmaspiegel konnten durch eine Ergänzung mit inaktivem Vitamin D wieder normalisiert werden.
Unerwünschte Wirkungen bei erwachsenen Patienten
Die häufigsten unerwünschten Arzneimittelwirkungen, die bei erwachsenen Patienten im Verlauf klinischer Studien berichtet wurden, waren Rückenschmerzen (15%), Kopfschmerzen (13%), Zahninfektion (13%), Restless-Legs-Syndrom (12%), erniedrigtes Vitamin D (12%) und Schwindel (10%).
Tabelle 5: Unerwünschte Wirkungen bei erwachsenen Patienten
|
Systemorganklasse
|
Sehr häufig
|
Häufig
| |
Infektionen und parasitäre Erkrankungen
|
Zahninfektion (13%)
|
| |
Stoffwechsel- und Ernährungsstörungen
|
Vitamin D-Mangel (12%)
|
Hyperphosphatämie
| |
Erkrankungen des Nervensystems
|
Kopfschmerzen (13%),
Restless-Legs-Syndrom (12%),
Schwindel (10%)
|
| |
Erkrankungen des Gastrointestinaltrakts
|
|
Obstipation
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Rückenschmerzen (15%)
|
Muskelspasmen
Spinalkanalstenose
|
Beschreibung spezifischer unerwünschter Wirkungen bei Erwachsenen und Zusatzinformationen
Reaktionen an der Injektionsstelle
Die Häufigkeit von Reaktionen an der Injektionsstelle (Reaktionen an der Injektionsstelle, Erythem, Ausschlag, Schmerzen, Pruritus und Hämatom) betrug sowohl in der Burosumab- als auch in der Placebo-Gruppe jeweils 12%. Die Reaktionen an der Injektionsstelle traten meist innerhalb von 1 Tag nach der Injektion des Arzneimittels auf, hielten etwa 1 bis 3 Tage an, erforderten keine Behandlung und klangen in fast allen Fällen wieder ab.
Überempfindlichkeitsreaktionen
Die Häufigkeit potentieller Überempfindlichkeitsreaktionen war in der mit Burosumab und der mit Placebo behandelten Gruppe vergleichbar. Alle entsprechenden Ereignisse waren von leichtem bis mittlerem Schweregrad.
Hyperphosphatämie
In der doppelblinden Phase der Studie UX023-CL303 trat bei 7% der Patienten in der Burosumab-Behandlungsgruppe eine die im Prüfplan spezifizierten Kriterien für eine Dosisreduktion erfüllende Hyperphosphatämie auf (entweder ein einzelner Serumphosphatwert von mehr als 1.615 mmol/l [5.0 mg/dl] oder ein Serumphosphatwert von mehr als 1.454 mmol/l [4.5 mg/dl] [Obergrenze des Normbereichs] bei zwei verschiedenen Messungen). Im Falle einer Hyperphosphatämie erfolgte eine Dosisreduktion um 50%. Bei einem Patienten war eine zweite Dosisreduktion wegen nach wie vor bestehender Hyperphosphatämie erforderlich.
Restless-Legs-Syndrom
Bei etwa 12% der Patienten unter Burosumab und 8% der Patienten unter Placebo kam es zu einer Verschlimmerung eines zu Studienbeginn vorhandenen Restless-Legs-Syndroms bzw. zum erstmaligen Auftreten eines Restless-Legs-Syndroms.
Immunogenität
Die Inzidenz von Anti-Drug-Antikörpern (ADA) gegen Burosumab lag insgesamt bei <10 % der Probanden, die Burosumab erhielten. Der Nachweis von Antikörpern war weder mit einer reduzierten Wirksamkeit noch mit einer Häufung unerwünschter Ereignisse oder mit Veränderungen des pharmakokinetischen Profils assoziiert.
Erniedrigte Vitamin D-Spiegel
Nach Einleitung der Behandlung mit Burosumab wurden bei ca. 12% der Patienten verminderte 25-Hydroxy-Vitamin D-Spiegel (inaktives Vitamin D) im Serum beobachtet, möglicherweise aufgrund einer erhöhten Umwandlung zu aktiviertem Vitamin D. Die Plasmaspiegel konnten durch eine Ergänzung mit inaktivem Vitamin D wieder normalisiert werden.
Spinalkanalstenose
Eine Spinalkanalstenose stellt bei Erwachsenen eine Komplikation des XLH dar, auch über Fälle mit Rückenmarkskompression wurde berichtet. In den Phase II/III-Studien bei Erwachsenen (n = 176) mussten sich insgesamt 6 Patienten einer Wirbelsäulenoperation unterziehen. Bei den meisten Fällen schien eine Progression einer vorbestehenden Spinalkanalstenose beteiligt zu sein. Es ist nicht bekannt, ob die Therapie mit Burosumab eine Spinalkanalstenose oder eine Rückenmarkskompression verschlimmern kann.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIm Fall einer Überdosierung sollte Burosumab abgesetzt und die biochemische Reaktion beobachtet werden.
Eigenschaften/WirkungenATC-Code
M05BX05
Wirkungsmechanismus
Burosumab ist ein rekombinanter humaner monoklonaler IgG1 Antikörper gegen den Fibroblasten-Wachstumsfaktor 23 (FGF23), hergestellt mittels rekombinanter DNA Technologie. Burosumab bindet an den FGF23 und hemmt dessen Aktivität. Durch die Hemmung von FGF23 steigert Burosumab die tubuläre Rückresorption von Phosphat aus der Niere und erhöht die Serumkonzentration von 1,25-Dihydroxy-Vitamin D.
Pharmakodynamik
Siehe oben.
Klinische Wirksamkeit
Klinische Wirksamkeit bei Kindern mit XLH
Studie UX023-CL301
In der pädiatrischen Studie UX023-CL301 wurden 61 Patienten im Alter von 1 bis 12 Jahren (56% weiblich; 44% männlich, Alter bei der ersten Dosis: Mittelwert [SD]: 6.3 [3.31] Jahre) randomisiert einer Behandlung mit Burosumab (n = 29) oder einer aktiven Kontrolle (n = 32; orales Phosphatpräparat und aktives Vitamin D) zugewiesen. Beim Eintritt in die Studie mussten alle Patienten seit mindestens 6 Monaten mit einem oralen Phosphatpräparat und aktivem Vitamin D behandelt worden sein. Von allen Patienten lagen röntgenologische Nachweise einer XLHbedingten Knochenerkrankung (Rachitis-Severity-Score ≥2) vor. Die Behandlung mit Burosumab wurde mit einer Dosis von 0.8 mg/kg alle 2 Wochen eingeleitet und bei unzureichendem Ansprechen laut Nüchtern-Serumphosphatwert auf 1.2 mg/kg gesteigert. Patienten, die in die aktive Kontrollgruppe randomisiert wurden, erhielten mehrere tägliche Dosen eines oralen Phosphatpräparats und von aktivem Vitamin D.
Primärer Wirksamkeitsendpunkt war die Veränderung des Rachitis-Schweregrades in Woche 40 gemäss RGI-C-Score (Radiographic Global Impression of Change; radiographischer Gesamteindruck der Veränderung) im Vergleich zwischen der Burosumab-Gruppe und der aktiven Kontrollgruppe.
Der RGI-C ist eine relative Beurteilungsskala zum Vergleich der Rachitis vor und nach einer Behandlung mithilfe einer 7 Punkte umfassenden Ordinalskala, welche die Veränderung bei den gleichen Anomalien beurteilt, die anhand des (unten beschriebenen) RSS bewertet wurden. Die Punktbewertungen (Scores) reichen von -3 (schwere Verschlechterung der Rachitis) bis +3 (vollständige Heilung der Rachitis).
Der Schweregrad der pädiatrischen Rachitis wurde mithilfe des RSS bestimmt, einer Bewertungsmethode für Röntgenaufnahmen basierend auf dem Grad der Ausfransung oder Becherung der Metaphyse und inwieweit die Wachstumsfuge betroffen ist. In Studie UX023-CL301 wurde der RSS anhand einer vorgegebenen Skala bewertet, die spezifische Anomalien von Hand- und Kniegelenken berücksichtigte.
Alle Patienten durchliefen mindestens 64 Wochen der randomisierten Behandlung. Bei keinem der Patienten wurde die Dosis reduziert, und bei 8 (28%) der mit Burosumab behandelten Patienten wurde die Dosis auf 1.2 mg/kg erhöht.
Primäre Wirksamkeitsergebnisse
Bei der Behandlung mit Burosumab wurde im Vergleich zur aktiven Kontrolle eine bessere Heilung der Rachitis in Woche 40 beobachtet. Diese Wirkung hielt bis Woche 64 an, wie aus Abbildung 1 hervorgeht.
Abbildung 1: RGI-C-Gesamtscore (Mittelwert ± SE) – Primärer Wirksamkeitsendpunkt in Woche 40 und 64 (Gesamtgruppe)
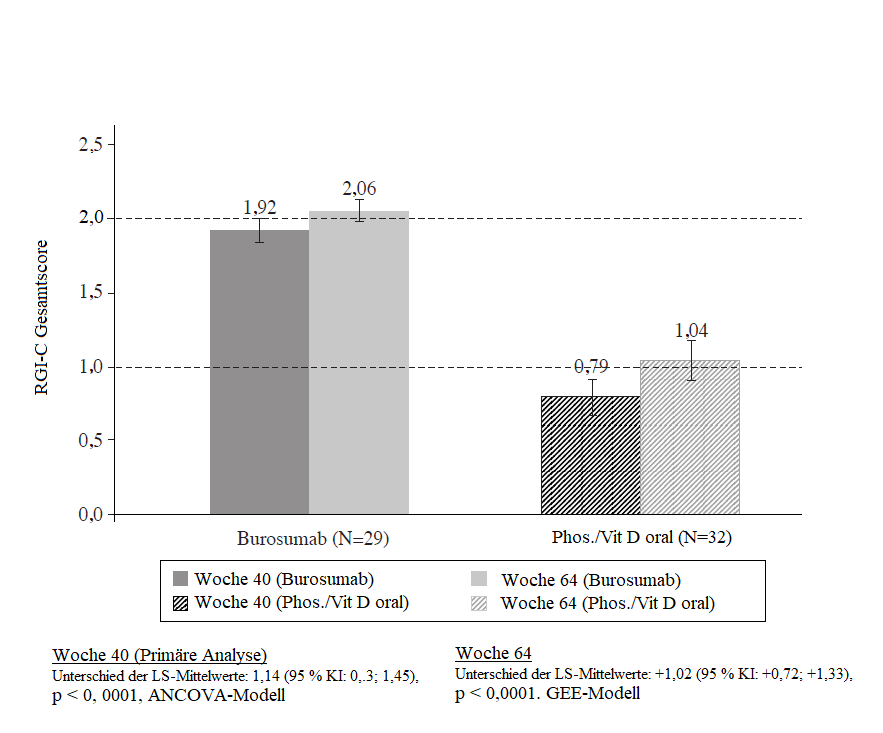
Sekundäre Wirksamkeitsergebnisse
Die wichtigsten Ergebnisse der sekundären Wirksamkeitsendpunkte sind in Tabelle 6 dargestellt.
Tabelle 6 Ergebnisse der sekundären Wirksamkeitsendpunkte
|
Endpunkt
|
Woche
|
Aktive Kontrolle
LS Mittelwert (SE)
|
Burosumab
LS Mittelwert (SE)
|
Unterschied (Burosumab – aktive Kontrolle)
| |
Missbildung der unteren Gliedmassen; gemäss RGI-C
(GEE-Modell)
|
40
|
+0.22 (0.080)
|
+0.62 (0.153)
|
+0.40 [95%-KI: 0.07, 0.72] p = 0.0162
| |
64
|
+0.29 (0.119)
|
+1.25 (0.170)
|
+0.97 [95%-KI: +0.57, +1.37] p < 0.0001
| |
Grösse; Z-Score
|
Studienbeginn
|
-2.05 (0.87)
|
-2.32 (1.17)
|
| |
40a
|
+0.03 (0.031)
|
+0.16 (0.052)
|
+0.12 [95%-KI: 0.01, 0.24] p = 0.0408
| |
64b
|
+0.02 (0.035)
|
+0.17 (0.066)
|
+0.14 [95%-KI: 0.00, 0.29] p = 0.0490
| |
Rachitis-Schweregrad, RSS-Gesamtscore
|
Studienbeginn
|
3.19 (1.141)
|
3.17 (0.975)
|
| |
40a
|
-0.72 (0.162)
|
-2.08 (0.104)
|
-1.34 [95%-KI: -1.74, -0.94] p < 0.0001
| |
64b
|
-1.01 (0.151)
|
-2.23 (0.117)
|
-1.21 [95%-KI: -1.59, -0.83] p < 0.0001
| |
Alkalische Phosphatase im Serum (U/L)
|
Studienbeginn
|
523 (154)
|
511 (125)
|
| |
40a
|
489 (189)
|
381 (99)
|
-97 [95% KI: -138, -56]
p < 0.0001
| |
64b
|
495 (182)
|
337 (86)
|
-147 [95% KI: -192, -102]
p < 0.0001
| |
Sechs-Minuten-Gehtest (m)
|
Studienbeginn
|
450 (106)
|
385 (86)
|
| |
40a
|
+4 (14)
|
+47 (16)
|
+43 [95%-KI: -0.3, 87]; p = 0.0514
| |
64b
|
+29 (17)
|
+75 (13)
|
+46 [95%-KI: 2, 89];
p = 0.0399
|
a Veränderung zwischen Studienbeginn und Woche 40 laut ANCOVA-Modell.
b Veränderung zwischen Studienbeginn und Woche 64 laut GEE-Modell.
Serumphosphat
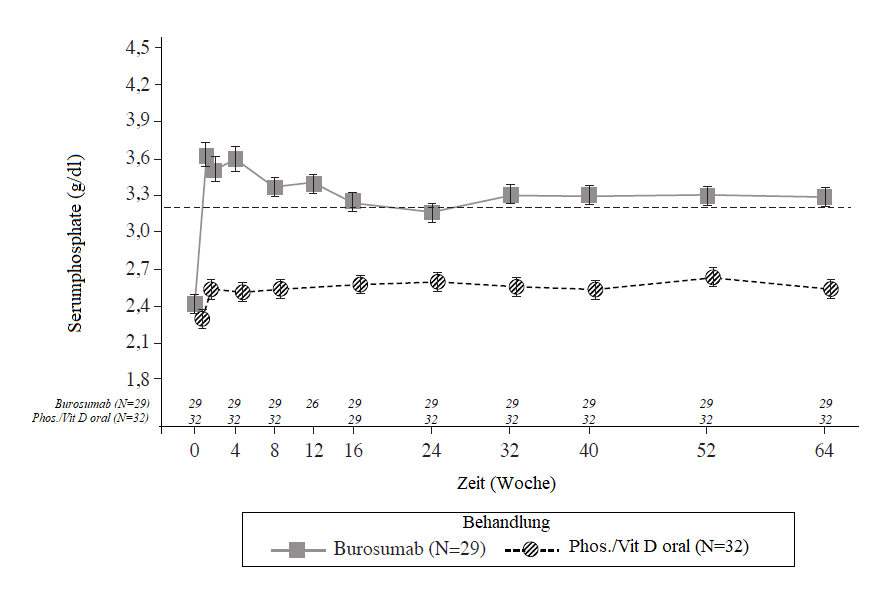
Bei jedem Studientermin, bei dem in beiden Gruppen die Serumphosphatwerte beurteilt wurden, waren die Veränderungen der Serumphosphatwerte gegenüber Studienbeginn in der Burosumab-Gruppe grösser als in der aktiven Kontrollgruppe (p < 0.0001; GEE-Modell) (Abbildung 2).
Abbildung 2: Serumphosphatkonzentration und Veränderung gegenüber Studienbeginn (mg/dl) (Mittelwert ± SE) nach Behandlungsgruppe (PD Analysegruppe)
Hinweis: Die gestrichelte Linie in der Abbildung zeigt die untere Grenze des Serumphosphat-Referenzbereichs von 3.2 mg/dl an (1.03 mmol/L).
Pädiatrische PROMIS-Fragebögen
In der Burosumab-Gruppe wurden in den Wochen 40 und 64 klinisch bedeutsame Verbesserungen in Bezug auf Schmerzbeeinträchtigung und Erschöpfung (erhoben für Patienten ≥ 5 Jahren) festgestellt, wobei zwischen den Behandlungsgruppen in Woche 40 ein statistisch signifikanter Unterschied zugunsten der Burosumab-Gruppe hinsichtlich der Schmerzbeeinträchtigung beobachtet wurde (p = 0.0212). In der aktiven Kontrollgruppe gab es in den Wochen 40 oder 64 keine klinisch bedeutsame Veränderung gegenüber den Ausgangswerten.
Studie UX023-CL201
In der pädiatrischen Studie UX023-CL201 wurden 52 Kinder im Alter von 5 bis 12 Jahren (mittleres Alter 8.5 Jahre; Standardabweichung (SD) 1.87) mit XLH über 64 Wochen behandelt. Bei fast allen Patienten lagen zu Studienbeginn röntgenologische Nachweise einer Rachitis vor, und sie waren zuvor über eine mittlere (SD) Zeitdauer von 7 (2.4) Jahren mit oralen Phosphatpräparaten und Vitamin D-Derivaten behandelt worden. Diese konventionelle Therapie wurde 2 bis 4 Wochen vor Einleitung der Burosumab-Behandlung abgesetzt. Die Burosumab-Dosis wurde an einen Zielwert für den Nüchtern-Serumphosphatwert von 3.50 bis 5.02 mg/dl (1.13 bis 1.62 mmol/l) angepasst. Von den 52 Patienten erhielten 26 Burosumab alle 4 Wochen (Q4W). Ebenfalls 26 der 52 Patienten erhielten Burosumab alle zwei Wochen (Q2W) in einer Durchschnittsdosis (Min., Max.) von 0.73 (0.3; 1.5), 0.98 (0.4; 2.0) und 1.04 (0.4; 2.0) mg/kg in Woche 16, 40 bzw. 64 und bis zu einer Maximaldosis von 2.0 mg/kg.
Burosumab erhöhte die Serumphosphat-Konzentration und den Quotienten aus maximaler tubulärer Rückresorption von Phosphat und glomerulärer Filtrationsrate (TmP/GFR-Quotienten). In der Patientengruppe, die Burosumab alle 2 Wochen erhielt, erhöhte sich die mittlere (SD) Serumphosphat-Konzentration von 2.38 (0.405) mg/dl (0.77 (0.13) mmol/l) zu Studienbeginn auf 3.30 (0.396) mg/dl (1.07 (0.13) mmol/l) in Woche 40 und blieb bis Woche 64 auf einem Wert von 3.35 (0.445) mg/dl (1.08 (0.14) mmol/l).
Aktivität der alkalischen Phosphatase
Die mittlere (SD) Gesamtaktivität der alkalischen Phosphatase im Serum betrug bei Ausgangswerterhebung 459 (105) E/l und sank auf 369 (76) E/l in Woche 64 (-19.6%; p <0.0001).
Der Gehalt an knochenspezifischer alkalischer Phosphatase im Serum betrug 165 (52) μg/l [Mittel (SD)] bei Ausgangswerterhebung und 115 (31) μg/l in Woche 64 (mittlere Veränderung: -28.5%).
Der Schweregrad der pädiatrischen Rachitis wurde in Studie UX023-CL201 mithilfe des RSS (siehe oben) bestimmt. In Studie UX023-CL201 wurde der RSS anhand einer vorgegebenen Skala bewertet, die spezifische Anomalien der Handgelenke und Knie berücksichtigte. Die Ergebnisse sind in Tabelle 7 zusammengefasst.
Als Ergänzung zur Beurteilung des RSS wurde die RGI-C Bewertungsskala verwendet. Die Ergebnisse sind in Tabelle 7 zusammengefasst.
Tabelle 7: Ansprechen der Rachitis bei Kindern von 5 bis 12 Jahren, die in Studie UX023-CL201 mit Burosumab behandelt wurden
|
Endpunkt
|
Dauer der Burosumab-Behandlung
(Woche)
|
Effektgrösse
| |
Alle 2 Wochen (N=26)
|
Alle 4 Wochen (N=26)
| |
RSS Gesamtscore
Mittelwert (SD) zu Studienbeginn
Veränderung des LS Mittelwertes (SE) des Gesamtscoresa gegenüber Baseline (ein reduzierter RSS-Score zeigt eine Besserung des Schweregrades der Rachitis)
|
|
|
| |
40
|
1.92 (1.2)
-1.06 (0.1) (p < 0.0001)
|
1.67 (1.0)
-0.73 (0.1) (p < 0.0001)
| |
64
|
-1.00 (0.1) (p < 0.0001)
|
-0.84 (0.1) (p < 0.0001)
| |
RGI-C Gesamtscore
LS Mittelwert (SE)a (positive weist auf Heilung hin)
|
40
|
+1.66 (0.1) (p < 0.0001)
|
+1.47 (0.1) (p < 0.0001)
| |
64
|
+1.56 (0.1) (p < 0.0001)
|
+1.58 (0.1) (p < 0.0001)
|
a Schätzungen des LS Mittelwerts und der p-Werte stammen von dem Gleichungsmodell für generalisierte Schätzungen, das den Ausgangs-RSS, Besuchstermine und Behandlungsregime sowie deren Interaktionen berücksichtigt.
Studie UX023-CL205
In der pädiatrischen Studie UX023-CL205 wurde Burosumab an 13 Kindern mit XLH im Alter von 1 bis 4 Jahren (mittleres Alter 2.9 Jahre; SD 1.1) über 40 Wochen untersucht. Bei allen Patienten lagen zu Studienbeginn röntgenologische Nachweise einer Rachitis vor, und zwölf Patienten waren für eine mittlere (SD) Zeitdauer von 16.7 (14.4) Monaten mit oralen Phosphatpräparaten und Vitamin D-Derivaten vorbehandelt worden. Diese konventionelle Therapie wurde 2 bis 6 Wochen vor Einleitung der Burosumab-Behandlung abgesetzt. Die Patienten erhielten Burosumab in einer Dosis von 0.8 mg/kg alle zwei Wochen.
In Studie UX023-CL205 stieg die mittlere (SD) Nüchtern-Serumphosphat-Konzentration von 2.51 (0.284) mg/dl (0.81 (0.092) mmol/l) zu Studienbeginn auf 3.47 (0.485) mg/dl (1.12 (0.158) mmol/l) in Woche 40.
Aktivität der alkalischen Phosphatase im Serum
Die mittlere (SD) Gesamtaktivität der alkalischen Phosphatase im Serum betrug 549 (193.8) E/l zu Studienbeginn und sank auf 335 (87.6) E/l in Woche 40 (mittlere Veränderung: -36.3%).
Rachitis-Severity-Score (RSS; Score zur Erfassung des Schweregrads der Rachitis)
Nach 40 Behandlungswochen mit Burosumab besserte sich der mittlere Gesamt-RSS von 2.92 (1.367) zu Studienbeginn auf 1.19 (0.522); dies entsprach einer Veränderung des angepassten Mittelwerts (SE) gegenüber dem Ausgangswert von -1.73 (0.132) (p < 0.0001).
Radiographic Global Impression of Change (RGI-C; Radiographischer Gesamteindruck der Veränderung)
Nach 40 Behandlungswochen mit Burosumab betrug der angepasste Mittelwert (SE) des RGI-C-Gesamtscores +2.33 (0.08) bei allen 13 Patienten (p < 0.0001), was eine Besserung der Rachitis belegte. Alle 13 Patienten wurden als RGI-C-Responder gemäss der Definition durch einen RGI-C-Gesamtscore ≥ +2.0 eingestuft.
Klinische Wirksamkeit bei Erwachsenen mit XLH
Studie UX023-CL303 ist eine randomisierte, doppelblinde, placebokontrollierte Studie an 134 erwachsenen XLH-Patienten. Die Studie umfasste eine 24-wöchige placebokontrollierte Behandlungsphase, gefolgt von einer 24-wöchigen offenen Phase, in der alle Patienten Burosumab erhielten. Burosumab wurde in einer Dosis von 1 mg/kg alle 4 Wochen verabreicht. Ein Patient in der Burosumab-Gruppe brach die Behandlung ab. Zu Studienbeginn lag das mittlere Alter der Patienten bei 40 Jahren (Spanne 19 bis 66 Jahre) und 35% waren männlich. Die mittlere (SE) Phosphatkonzentration im Serum zu Studienbeginn lag unterhalb der unteren Normgrenze bei 1.98 (0.314) mg/dl (0.64 (1.10) mmol/l).
In Studie UX023-CL304 wurde der Einfluss von Burosomab auf die durch die Hypophosphatämie bedingte Osteomalazie anhand der Untersuchung von Beckenkamm-Biopsien evaluiert. Es handelte sich um eine 48-wöchige, offene, einarmige Studie an 14 erwachsenen Patienten mit XLH. Die Patienten erhielten alle 4 Wochen 1.0 mg/kg Burosumab.
Serumphosphat
In Studie UX023-CL303 lag der Serumphosphatwert zu Studienbeginn bei 1.92 (0.32) mg/dl (0.62 (0.102) mmol/l) in der Placebo- und bei 2.03 (0.30) mg/dl (0.66 (0.098) mmol/l in der Burosumab-Gruppe. Im Verlauf der 24 Behandlungswochen betrug der mittlere (SD) Serumphosphatwert in der Mitte der Verabreichungsintervalle (2 Wochen nach Verabreichung) 2.08 (0.30) mg/dl (0.67 (0.09) mmol/l) in der Placebo- bzw. 3.24 (0.53) mg/dl (1.05 (0.17) mmol/l) in der Burosumab-Gruppe. Für den primären Wirksamkeitsendpunkt sind die Resultate in Tabelle 8 aufgeführt.
Tabelle 8: Anteil der erwachsenen Patienten, die in Studie UX023-CL303 (doppelblinde Phase) in der Mitte des Verabreichungsintervalls einen mittleren Serumphosphatwert über der LLN erreichten
|
|
Placebo
(N = 66)
|
Burosumab
(N = 68)
| |
In der Mitte der Verabreichungsintervalle bis Woche 24 ein mittleres Serumphosphat > LLN erreicht - n (%)
|
7.6% (5/66)
|
94.1% (64/68)
| |
95%-KI
|
(3.3, 16.5)
|
(85.8, 97.7)
| |
p-Werta
|
|
< 0.0001
|
Die 95%-KIs wurden mithilfe der Wilson-Score-Methode berechnet.
a Der P-Wert stammt aus einem Cochran-Mantel-Haenszel (CMH) Test auf Assoziation zwischen dem Erreichen des primären Endpunkts und der Behandlungsgruppe zur Adjustierung für die Stratifizierungen der Randomisierung.
Die drei wichtigsten sekundären, von den Patienten berichteten Endpunkte Steifigkeit, stärkster Schmerz (Worst Pain) und körperliche Funktion (Physical Function) wurden in Woche 24 ausgewertet und mit den Ausgangswerten verglichen. Die Ergebnisse sind in Tabelle 9 dargestellt.
Tabelle 9: Veränderung der von den Patienten berichteten Ergebnisse zwischen Studienbeginn und Woche 24 sowie Auswertung des Unterschieds in Woche 24
|
|
Placebo
(N = 66)
|
Burosumab
(N = 68)
| |
WOMAC-Steifigkeitsscore
(Abnahme bedeutet geringere Steifigkeit)
|
|
| |
Mittelwert (SD) zu Studienbeginn
|
61.36 (20.77)
|
64.71 (20.25)
| |
Mittelwert (SD) in Woche 24
|
60.38 (21.83)
|
53.73 (20.76)
| |
Veränderung des LS Mittelwertes (SE) zwischen Studienbeginn und Woche 24
|
+0.46 (3.14)
|
-7.85 (3.03)
| |
Unterschied im LS Mittelwert (SE) (Burosumab vs. Placebo)
|
-8.3 (3.25)
| |
p-Wert
|
0.0106
| |
WOMAC Physical Function-Score
(Abnahme bedeutet Besserung der körperlichen Funktionsfähigkeit)
|
|
| |
Mittelwert (SD) zu Studienbeginn
|
43.89 (19.94)
|
50.79 (19.66)
| |
Mittelwert (SD) in Woche 24
|
42.65 (22.76)
|
43.43 (19.51)
| |
Veränderung des LS Mittelwertes (SE) zwischen Studienbeginn und Woche 24
|
+1.79 (2.72)
|
-3.11 (2.55)
| |
Unterschied (SE) (Burosumab vs. Placebo)
|
-4.9 (2.48)
| |
p-Wert
|
0.0478
| |
BPI-SF Worst Pain-Score
(Abnahme bedeutet weniger Schmerz)
|
|
| |
Mittelwert (SD) zu Studienbeginn
|
6.54 (1.43)
|
6.81 (1.31)
| |
Mittelwert (SD) in Woche 24
|
6.09 (2.01)
|
5.82 (1.92)
| |
Veränderung des LS Mittelwertes (SE) zwischen Studienbeginn und Woche 24
|
-0.32 (0.22)
|
-0.79 (0.21)
| |
Unterschied (SE) (Burosumab vs. Placebo)
|
-0.5 (0.28)
| |
p-Werta
|
0.0919
| |
BPI-SF = Brief Pain Inventory (Schmerzfragebogen); GEE = generalized estimation equation (generalisierte Schätzgleichung); LS = least squares (kleinste Quadrate); WOMAC = Western Ontario and McMaster Universities osteoarthritis index (Arthroseindex der Universitäten Western Ontario und McMaster)
a Schätzungen der LS-Mittelwerte und der p-Werte in Woche 24 stammen von dem Gleichungsmodell für generalisierte Schätzungen, das die Veränderung seit Studienbeginn für den Endpunkt von Interesse als abhängige Variable, Region, Besuchstermin, Behandlungsregime, tatsächliche Stratifizierung bei Randomisierung (nicht bei der Auswertung des BPI Worst Pain-Scores) und die Interaktion zwischen Besuchstermin und Behandlungsregime als Festfaktoren sowie den Ausgangswert für den Endpunkt von Interesse als Kovariable mit einer zusammengesetzt-symmetrischen Kovarianzstruktur berücksichtigt.
|
Radiologische Beurteilung von Frakturen und Pseudofrakturen
In Studie UX023-CL303 wurde zu Studienbeginn ein Skelettröntgen durchgeführt, um osteomalaziebedingte Frakturen und Pseudofrakturen zu identifizieren. Zu Studienbeginn hatten 52% der Patienten entweder aktive (nicht verheilte) Frakturen (12%) oder aktive Pseudofrakturen (47%). In Woche 24 waren 36.9% der zu Studienbeginn aktiven Frakturen/Pseudofrakturen in der Burosumab-Gruppe vollständig abgeheilt, verglichen mit 9.9% in der Placebo-Gruppe.
Histomorphometrische Untersuchungen der Knochenstruktur bei Erwachsenen
Nach 48 Behandlungswochen in Studie UX023-CL304 wurde bei 10 Patienten anhand von Abnahmen des Osteoidvolumens/Knochenvolumens (OV/BV) von einem mittleren (SD) Score von 26.1% (12.4) bei Studienbeginn auf 11.2% (6.5), einer Reduktion um 57%, eine Heilung der Osteomalazie beobachtet.
PharmakokinetikDie folgenden pharmakokinetischen Parameter wurden, soweit nicht anders angegeben, bei Patienten mit XLH erhoben, die die zugelassene empfohlene Anfangsdosis – bezogen auf einen Patienten mit einem Körpergewicht von 70 kg – erhielten.
Burosumab zeigte nach Injektion s.c. in Dosen innerhalb einer Spanne von 0.1 bis 1 mg/kg (dem 0.08- bis 0.8-fachen der zugelassenen empfohlenen Höchstdosis, bezogen auf einen Patienten mit einem Körpergewicht von 70 kg) eine lineare Pharmakokinetik. Die mittlere (± SD) Talkonzentration von Burosumab im Steady-State belief sich bei erwachsenen Patienten auf 5.8 (± 3.4) µg/ml.
Absorption
Die Absorption von Burosumab in den Blutkreislauf ist nach subkutaner Gabe nahezu vollständig. Die mittleren Tmax-Werte nach Verabreichung von Burosumab bewegten sich in einem Bereich zwischen 8 und 11 Tagen.
Distribution
Das scheinbare Distributionsvolumen von Burosumab beträgt 8 l.
Metabolismus
Eine genaue Beschreibung des Stoffwechselwegs, auf dem Burosumab metabolisiert wird, liegt nicht vor. Es ist davon auszugehen, dass Burosumab über katabole Wege zu kleinen Peptiden und Aminosäuren abgebaut wird.
Elimination
Die Clearance von Burosumab ist abhängig vom Körpergewicht und liegt bei schätzungsweise 0.290 L/Tag bei einem typischen Erwachsenen (70 kg) bzw. 0.136 L/Tag bei einem typischen pädiatrischen Patienten (30 kg) mit XLH, wobei die entsprechende Dispositions-Halbwertszeit (t1/2) im Serum bei etwa 19 Tagen liegt. Nach mehrfacher Dosisgabe erreichen die Serumtalkonzentrationen 8 Wochen nach Behandlungsbeginn ein Plateau.
Linearität/Nicht Linearität
Burosumab zeigt eine zeitinvariante, dosislineare Pharmakokinetik über den subkutanen Dosisbereich von 0.1 bis 1.0 mg/kg.
Pharmakokinetischer/pharmakodynamischer Zusammenhang
Bei subkutaner Anwendung ist ein direkter pharmakokinetischer/pharmakodynamischer Zusammenhang zwischen den Burosumab-Serumkonzentrationen und den Anstiegen der Serumphosphat-Konzentration zu beobachten, der durch ein Emax/EC50-Modell gut dargestellt wird. Die Burosumab- und Phosphat-Serumkonzentrationen sowie TmP/GFR stiegen und sanken parallel und erreichten Höchstkonzentrationen zu etwa dem gleichen Zeitpunkt nach jeder Dosis, was auf einen direkten pharmakokinetischen/pharmakodynamischen Zusammenhang hindeutet. Die AUC für die Veränderung von Serumphosphat, TmP/GRF und 1,25-Dihydroxy-Vitamin D gegenüber dem jeweiligen Ausgangswert stiegen mit einer ansteigenden Burosumab-AUC linear an.
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
In Bezug auf die Pharmakokinetik oder Pharmakodynamik bei Kindern und Jugendlichen wurde kein signifikanter Unterschied zur Pharmakokinetik/Pharmakodynamik bei Erwachsenen festgestellt. Clearance und Verteilungsvolumen von Burosumab sind vom Körpergewicht abhängig. Es wurden keine klinisch bedeutsamen altersabhängigen Unterschiede bei der Pharmakokinetik von Burosumab beobachtet.
Leber- und Nierenfunktionsstörungen
Die Auswirkungen einer eingeschränkten Nieren- oder Leberfunktion auf die Pharmakokinetik von Burosumab wurden nicht untersucht.
Präklinische DatenSicherheitspharmakologie
In präklinischen Studien an gesunden Tieren wurden nach Expositionen, die zu Serumphosphat-Konzentrationen über den Normwerten führten, unerwünschte Wirkungen beobachtet. Diese Wirkungen stimmten mit einer übersteigerten Reaktion auf die Hemmung normaler FGF23-Spiegel überein, die zu einem supraphysiologischen Anstieg des Serumphosphats über den oberen Normwert hinausführten.
Studien an Kaninchen und erwachsenen sowie juvenilen Makakenaffen zeigten dosisabhängige Anstiege von Serumphosphat und 1,25-Dihydroxy-Vitamin D, welche die pharmakologischen Wirkungen von Burosumab bei diesen Tierspezies bestätigten. Bei gesunden Tieren wurde nach Burosumab-Dosen, die zu Serumphosphat-Konzentrationen über ca. 8 mg/dl (2.6 mmol/l) bei den Tieren führten, ektopische Mineralisierungen in verschiedenen Geweben und Organen (z. B. Niere, Herz, Lunge und Aorta) und in manchen Fällen damit assoziierte Sekundärerkrankungen (z. B. Nephrokalzinose) infolge von Hyperphosphatämie beobachtet. Diese Befunde wurden bei Expositionen die äquivalent oder niedriger als der Expositionsbereich in der empfohlenen klinischen Dosierung festgestellt. In einem murinen XLH-Modell wurde eine signifikante Reduktion der Inzidenz von ektopischen Mineralisierungen bei äquivalenten Serumphosphatspiegeln beobachtet, was darauf schliessen lässt, dass das Risiko für Mineralisierungen in Gegenwart einer übermässig grossen Menge an FGF23 geringer ist.
Knochenveränderungen wurden bei erwachsenen und juvenilen Affen beobachtetet (Veränderungen von Knochenstoffwechsel-Markern sowie einer Zunahme der Gesamtknochendichte und Verdickung der Röhrenknochen). Diese Befunde waren eine Folge der über den Normwert hinaus erhöhten Serumphosphatspiegel, die bei den untersuchten Dosen zu einer Beschleunigung des Knochenumsatzes und auch zu periostaler Hyperostose sowie einer Abnahme der Knochenfestigkeit bei erwachsenen Tieren, jedoch nicht bei juvenilen Tieren führten. Burosumab begünstigte keine abnormale Knochenentwicklung, da bei juvenilen Tieren keine Veränderungen der Femurlänge oder Knochenfestigkeit festzustellen waren. Die Knochenveränderungen waren mit der Pharmakologie von Burosumab und der Rolle, die Phosphat für die Knochenmineralisierung, den Knochenstoffwechsel und den Knochenumsatz spielt, vereinbar.
Toxizität bei wiederholter Gabe
In Studien zur Toxikologie nach mehrmaliger Anwendung mit einer Dauer von bis zu 40 Wochen an Makakenaffen wurde bei männlichen Affen eine Mineralisierung des Rete testis/der Samenkanälchen beobachtet. Bei der Analyse der Samenflüssigkeit wurden jedoch keine Veränderungen festgestellt. In diesen Studien wurden keine unerwünschten Wirkungen auf die Fortpflanzungsorgane der weiblichen Tiere beobachtet.
Reproduktionstoxizität
Es wurden keine Tierexperimentelle Studien zur Fertilität mit Burosumab durchgeführt.
In den Studien zur Fortpflanzungs- und Entwicklungstoxizität in trächtigen Affen wurde bei einer Exposition, die dem 64-fachen der humanen Exposition in der klinischen Dosierung entspricht, eine Serumphosphat-Höchstkonzentration von über ca. 8 mg/dl (2.6 mmol/l) und eine moderate Mineralisierung der Plazenta beobachtet. Nach Dosen von ≥ 0.3 mg/kg, die einer Burosumab-Exposition vom ≥0.875- bis 1.39-Fachen der erwarteten klinischen Exposition entsprachen, wurden eine Verkürzung der Trächtigkeitsdauer und eine damit verbundene erhöhte Inzidenz von vorzeitigen Würfen bei trächtigen Affenweibchen beobachtet. Burosumab wurde im Serum von Föten nachgewiesen, was darauf hindeutet, dass Burosumab die Plazentaschranke passiert und auf den Fötus übergeht. Es gab keine Anzeichen für teratogene Wirkungen. Bei Föten oder bei den Nachkommen wurde keine ektopische Mineralisierung beobachtet. Burosumab hatte keinen Einfluss auf das prä- und postnatale Wachstum oder die Überlebensfähigkeit der Nachkommen.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Im Kühlschrank (2-8°C) lagern. Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Hinweise für die Handhabung
Klare bis leicht opaleszierende, farblose bis schwach bräunlich-gelbe Lösung.
Jede Durchstechflasche ist nur zum einmaligen Gebrauch bestimmt.
Durchstechflasche vor der Anwendung nicht schütteln.
Zulassungsnummer66801 (Swissmedic).
PackungenCRYSVITA 10 mg/ml Injektionslösung: Packung mit 1 Durchstechflasche mit 10 mg Burosumab in 1 ml Lösung. (B)
CRYSVITA 20 mg/ml Injektionslösung: Packung mit 1 Durchstechflasche mit 20 mg Burosumab in 1 ml Lösung. (B)
CRYSVITA 30 mg/ml Injektionslösung: Packung mit 1 Durchstechflasche mit 30 mg Burosumab in 1 ml Lösung. (B)
ZulassungsinhaberinKyowa Kirin Sàrl, Genève
Stand der InformationAugust 2024
|