Zusammensetzung
Wirkstoffe
Benralizumab.
Benralizumab ist ein humanisierter monoklonaler Antikörper, der mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters hergestellt wird.
Hilfsstoffe
L-Histidin, L-Histidin-Hydrochloridmonohydrat, α,α-Trehalose-Dihydrat, Polysorbat 20, Wasser für Injektionszwecke.
Indikationen/Anwendungsmöglichkeiten
Fasenra ist als Zusatz zur Erhaltungstherapie bei erwachsenen Patienten ab 18 Jahren mit schwerem eosinophilem Asthma indiziert, welches durch folgende Kriterien gekennzeichnet ist:
·mindestens zwei Exazerbationen in den vorausgegangenen 12 Monaten unter aktueller Standardtherapie (hochdosierte inhalative Kortikosteroide plus langwirksame Bronchodilatatoren) und/oder Notwendigkeit zur Behandlung mit systemischen Kortikosteroiden.
·Eosinophilenzahl im Blut von ≥0,3 G/Liter (entspricht ≥300 Zellen/μl).
Für genauere Angaben zu den in Studien untersuchten Patientenpopulationen siehe «Klinische Wirksamkeit».
Dosierung/Anwendung
Fasenra soll von einem Arzt verschrieben werden, der Erfahrung in der Diagnose und Behandlung von schwerem Asthma hat.
Übliche Dosierung
Die empfohlene Dosis beträgt 30 mg und wird für die ersten drei Dosen einmal alle 4 Wochen und danach alle 8 Wochen subkutan verabreicht.
Nach spätestens 5 Gaben Fasenra sollte der Therapieerfolg beurteilt und über die Fortführung der Behandlung entschieden werden. Zur Beurteilung des Ansprechens auf die Zusatztherapie sind eine sorgfältige Erfassung der Asthmakontrolle, des Bedarfs an systemischen Kortikosteroiden und der Exazerbationshäufigkeit vor und unter der Behandlung notwendig. Im Falle eines Ansprechens ist Fasenra für die Langzeitbehandlung vorgesehen. Eine Entscheidung über die Fortsetzung der Therapie sollte jeweils mindestens jährlich, basierend auf dem Schweregrad der Erkrankung und dem Grad der Kontrolle der Exazerbation getroffen werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Patienten mit Leberfunktionsstörungen
Bei Patienten mit eingeschränkter Leberfunktion ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit eingeschränkter Nierenfunktion ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Ältere Patienten
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Bei Jugendlichen unter 18 Jahren mit schwerem eosinophilem Asthma liegen beschränkte Daten vor. Eine Anwendung in dieser Altersgruppe wird nicht empfohlen.
Art der Anwendung
Fasenra sollte als subkutane Injektion initial von einem Arzt oder einer medizinischen Fachperson verabreicht werden. Die Injektion erfolgt in den Oberschenkel oder das Abdomen.
Wenn es der Arzt für angemessen hält, können Patienten, die Fasenra bereits wiederholt gut vertragen haben, sich das Präparat nach sorgfältiger entsprechender Instruktion entweder selbst injizieren oder sich von instruierten Betreuungspersonen injizieren lassen (siehe Gebrauchsanweisung am Ende der Patienteninformation). Wenn jemand anderes die Injektion verabreicht, kann die Injektion auch in den Oberarm erfolgen.
Fasenra soll nicht in Bereichen verabreicht werden, in denen die Haut empfindlich oder verhärtet ist oder sich ein Erythem oder Hämatom befindet (siehe «Sonstige Hinweise»).
Gemäss gängiger klinischer Praxis wird nach der Verabreichung eines biologischen Wirkstoffs eine Überwachung der Patienten empfohlen (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und Vorsichtsmassnahmen
Fasenra sollte nicht zur Behandlung akuter Asthma-Exazerbationen eingesetzt werden.
Die Patienten sollten angewiesen werden ärztliche Hilfe in Anspruch zu nehmen, wenn ihr Asthma nach Beginn der Behandlung weiterhin unkontrolliert ist oder sich verschlechtert.
Das abrupte Absetzen von Kortikosteroiden nach Beginn der Behandlung mit Fasenra wird nicht empfohlen. Die Dosisverringerung der Kortikosteroide sollte unter Aufsicht eines Arztes und stufenweise erfolgen.
Überempfindlichkeitsreaktionen
Nach der Verabreichung von Fasenra sind Überempfindlichkeitsreaktionen (z.B. Anaphylaxie, Angioödem, Urtikaria, papulöse Urtikaria, Hautausschlag) aufgetreten. Diese Reaktionen können in der Regel innerhalb der ersten Stunden nach der Verabreichung auftreten, in manchen Fällen jedoch auch verzögert (d.h. nach mehreren Tagen). Patienten müssen über dieses Risiko und die Behandlungsmöglichkeiten schwerer allergischer Reaktionen informiert werden.
Im Falle einer Überempfindlichkeitsreaktion sollte die Behandlung mit Fasenra abgebrochen werden und eine geeignete Therapie eingeleitet werden.
Bei Selbst-Anwendung sollten Patienten angewiesen werden, bei schweren systemischen allergischen Reaktionen wie Urtikaria, Angioödem, Atemschwierigkeiten oder Herz-Kreislaufstörungen sofort einen Arzt zu konsultieren.
Parasitäre Infektionen (Helminthen)
Eosinophile können an der Immunantwort auf einen Befall mit bestimmten Helminthen beteiligt sein. Patienten mit einer bekannten Helmintheninfektion wurden von den klinischen Studien ausgeschlossen. Es ist nicht bekannt, ob Fasenra die Reaktion des Patienten auf Helmintheninfektionen beeinflusst.
Patienten, bei denen ein Helminthenbefall vorliegt, sollten vor der Verabreichung von Fasenra entsprechend behandelt werden. Bei Patienten, bei denen während der Behandlung mit Fasenra ein Helminthenbefall eintritt, und die nicht auf eine Behandlung gegen Helminthen ansprechen, soll die Behandlung mit Fasenra unterbrochen werden, bis die Helmintheninfektion ausgeheilt ist.
Interaktionen
Es wurden keine formalen Wechselwirkungsstudien mit Benralizumab und anderen Arzneimitteln durchgeführt.
In einer randomisierten, doppelblinden, Parallelgruppenstudie mit 103 Patienten im Alter zwischen 12 und 21 Jahren mit schwerem Asthma, variierte die nach einer saisonalen Grippeimpfung gemessene humorale Antikörperreaktion abhängig vom Impfstamm und Messendpunkt. Bei hoher Variabilität zeigten sich keine konsistenten Unterschiede zwischen Placebo und Benralizumab.
Schwangerschaft, Stillzeit
Schwangerschaft
Es liegen keine aussagekräftigen Daten zur Anwendung von Benralizumab während der Schwangerschaft vor.
Tierexperimentelle Studien geben keine Hinweise auf eine Reproduktionstoxizität (siehe «Präklinische Daten»).
Monoklonale Antikörper, wie z.B. Benralizumab, werden im Verlauf der Schwangerschaft linear über die Plazenta transportiert. Aus diesem Grund ist eine potentielle Exposition des ungeborenen Kindes im zweiten und dritten Trimester der Schwangerschaft wahrscheinlich grösser.
Während der Schwangerschaft darf Fasenra nicht verabreicht werden, es sei denn dies ist eindeutig erforderlich.
Stillzeit
Es ist nicht bekannt, ob Benralizumab in die menschliche oder tierische Milch übergeht, daher kann ein Risiko für einen Säugling nicht ausgeschlossen werden.
Unter Abwägung der Vorteile des Stillens für das Kind und des Nutzens der Behandlung für die Mutter muss entweder das Stillen unterbrochen werden oder auf die Behandlung mit Benralizumab verzichtet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine entsprechenden Studien durchgeführt.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
In klinischen Studien mit Patienten mit schwerem unkontrolliertem eosinophilem Asthma waren die am häufigsten berichteten unerwünschten Wirkungen während der Behandlung Kopfschmerzen und Pharyngitis.
Auflistung der unerwünschten Arzneimittelwirkungen
Insgesamt erhielten 1'663 Patienten mit schwerem unkontrolliertem eosinophilem Asthma Benralizumab in klinischen Studien von 48- bis 56-wöchiger Dauer. Die folgenden unerwünschten Arzneimittelwirkungen wurden in 2 Placebo-kontrollierten Studien mit Patienten, die für die ersten 3 Dosen alle 4 Wochen und danach alle 8 Wochen 30 mg Benralizumab erhielten, festgestellt.
Die Häufigkeiten sind folgendermassen definiert: Sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1'000, < 1/100), selten (≥1/10'000, < 1/1'000), sehr selten (< 1/10'000) und nicht bekannt (Häufigkeit kann auf Basis der vorhandenen Daten nicht angegeben werden). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach absteigendem Schweregrad geordnet.
Erkrankung des Nervensystems
Häufig: Kopfschmerzen.
Infektionen und parasitäre Erkrankungen
Häufig: Pharyngitis*.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Fieber, Reaktionen an der Injektionsstelle.
Erkrankungen des Immunsystems
Häufig: Überempfindlichkeitsreaktion**.
Nicht bekannt: Anaphylaxie (einschliesslich Anaphylaktische Reaktion), Angioödem ***.
* Pharyngitis schliesst folgende Standardbegriffe ein: Pharyngitis, bakterielle Pharyngitis, virale Pharyngitis, Pharyngitis durch Streptokokken
** Überempfindlichkeitsreaktion schliesst folgende Standardbegriffe ein: Urtikaria, papulöse Urtikaria und Hautausschlag. Für Beispiele von berichteten assoziierten Manifestationen und der möglichen Zeit bis zum Einsetzen siehe «Warnhinweise und Vorsichtsmassnahmen».
*** Es handelt sich um eine unerwünschte Wirkung, die nach der Zulassung in Zusammenhang mit der Anwendung von Fasenra beobachtet wurde. Es ist generell nicht möglich, die Häufigkeit dieser Wirkungen zu bestimmen, da es sich um Spontanmeldungen aus einer Population nicht genau bekannter Grösse handelt. Die Häufigkeit dieser unerwünschten Wirkungen wird daher mit «nicht bekannt» (auf Grundlage der verfügbaren Daten nicht abschätzbar) angegeben.
Beschreibung ausgewählter Nebenwirkungen
Reaktionen an der Injektionsstelle
In Placebo-kontrollierten Studien traten bei 2,2% der mit Benralizumab behandelten Patienten Reaktionen an der Injektionsstelle (z.B. Schmerzen, Erythem, Juckreiz, Papeln) auf, im Gegensatz zu 1,9% der mit Placebo behandelten Patienten.
Langzeitsicherheit
In der BORA-Studie (Studie 4), eine 56-wöchige doppelblinde, randomisierte Extensionsstudie mit Asthmapatienten aus den Studien 1, 2 und 3, wurden 842 Patienten mit Fasenra mit der genehmigten Dosierung behandelt. Das Nebenwirkungsprofil entsprach dem in den oben genannten Asthma-Studien beschriebenen Nebenwirkungsprofil.
Die Studie 5 (MELTEMI) war eine offene Extensionsstudie zur Untersuchung der Sicherheit, in die erwachsene Patienten aufgenommen wurden, die mindestens 16 Wochen in Studie 4 behandelt worden waren. Insgesamt wurden 226 Patienten bis zu 43 Monate lang mit Fasenra in der empfohlenen Dosierung behandelt. Zusammen mit der Behandlungsdauer aus vorherigen Studien entspricht dies einer medianen Beobachtungszeit von 3,4 Jahren (Bereich: 8,5 Monate – 5,3 Jahre). Das Sicherheitsprofil während dieser Beobachtungszeit entsprach dem bekannten Sicherheitsprofil von Fasenra.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Im Rahmen von klinischen Studien wurden Patienten mit eosinophilem Asthma Einzeldosen bis zu 200 mg subkutan verabreicht, ohne dass Hinweise auf dosisabhängige Toxizitäten erkennbar waren.
Es gibt keine spezifische Behandlung für eine Überdosierung mit Benralizumab. Im Falle einer Überdosierung muss der Patient die jeweils angemessene unterstützende Behandlung erhalten und entsprechend überwacht werden.
Eigenschaften/Wirkungen
ATC-Code
R03DX10
Wirkungsmechanismus
Bei Benralizumab handelt es sich um einen humanisierten, afukosylierten, monoklonalen Antikörper (IgG1, Kappa). Benralizumab bindet mit hoher Affinität (16 pM) und Spezifität an die Alpha-Untereinheit des humanen Interleukin-5-Rezeptors (IL-5Rα). Der IL-5-Rezeptor wird besonders auf den Oberflächen von eosinophilen und basophilen Granulozyten exprimiert. Das Fehlen von Fukose im Fc-Teil von Benralizumab führt zu einer Bindung mit hoher Affinität (45,5 nM) an FcγRIII-Rezeptoren auf Immuneffektorzellen wie beispielsweise den natürlichen Killerzellen (NK). Diese Bindung führt zu einer verstärkten, antikörperabhängigen zellvermittelten Zytotoxizität (ADCC) und folglich zur Apoptose von Eosinophilen und Basophilen.
Die eosinophile Entzündung trägt bei eosinophilen Asthmaformen wesentlich zur Pathogenese der Erkrankung bei.
Pharmakodynamik
Wirkung auf Eosinophile im Blut
Die Behandlung mit Benralizumab führt innerhalb von 24 Stunden nach der ersten Dosis zu einer beinahe kompletten Depletion der Eosinophilen im Blut, welche während der gesamten Behandlungsperiode erhalten bleibt. Die Depletion der Eosinophilen im Blut wird begleitet von einer Reduktion von eosinophilen Granula-Proteinen im Serum, Eosinophil-derived Neurotoxin (EDN), und dem eosinophilen kationischen Protein (ECP) sowie einer Reduktion der Basophilen im Blut.
In den klinischen Studien 1 (SIROCCO) und 2 (CALIMA) verminderte sich nach der subkutanen Verabreichung der empfohlenen Benralizumab-Dosis die Eosinophilenanzahl im Blut auf einen medianen Absolutwert von 0 Zellen/μl, was einer medianen Verringerung von 100% entspricht (siehe «Klinische Wirksamkeit»). Dieses Ausmass der Verminderung wurde beim ersten Beobachtungszeitpunkt sowie nach 4 Wochen Behandlung festgestellt und blieb während des gesamten Behandlungszeitraums erhalten.
Über die gesamte 56-wöchige Extensionsphase (Studie 4, BORA) hinweg wurde eine Aufrechterhaltung der Eosinophilen-Depletion beobachtet, was in Einklang mit Befunden aus den früheren Studien steht.
Wirkung auf Eosinophile in der Atemwegsschleimhaut
Die Wirkung von Benralizumab auf Eosinophile der Atemwegsschleimhaut von Asthmapatienten mit erhöhter Anzahl Eosinophiler im Sputum (≥2,5%) wurde in einer 12wöchigen, Phase-1, randomisierten, doppelblinden, Placebo-kontrollierten, klinischen Studie mit Benralizumab 100 oder 200 mg subkutan evaluiert. In dieser Studie gab es eine mediane Verminderung der Eosinophilen in der Atemwegsschleimhaut gegenüber der Baseline um insgesamt 96% in der mit Benralizumab behandelten Gruppe im Vergleich zur Verringerung um 47% in der Placebo-Gruppe (p=0,039).
Klinische Wirksamkeit
Die Wirksamkeit von Fasenra wurde in 3 randomisierten, doppelblinden, Placebo-kontrollierten, klinischen Studien von 28- bis 56-wöchiger Dauer an Patienten ab 12 Jahren untersucht.
In diesen Studien wurde Fasenra mit einer Dosis von 30 mg einmal alle 4 Wochen für die ersten 3 Dosen und anschliessend alle 4 oder 8 Wochen als Zusatztherapeutikum zur Standardbehandlung verabreicht und vergleichend zur Standardbehandlung mit Placebo untersucht.
Die beiden Placebo-kontrollierten Exazerbationsstudien Studie 1 (SIROCCO) und Studie 2 (CALIMA) hatten eine Dauer von 48 bzw. 56 Wochen und es nahmen insgesamt 2'510 Patienten (Erwachsene oder Jugendliche ab 12 Jahren) mit unkontrolliertem Asthma teil. Davon wurden 822 Patienten mit der beantragten Dosis behandelt. In der Krankengeschichte der Patienten mussten in den vergangenen 12 Monaten mindestens 2 Asthma-Exazerbationen aufgetreten sein, die eine orale oder systemische Kortikosteroidbehandlung erforderten. Zudem musste eine ACQ-6 Auswertung von 1,5 oder höher beim Screening, sowie eine verringerte Lungenfunktion bei Baseline (vor Bronchodilatation forciertes Expirationsvolumen in 1 Sekunde (FEV1) < 80% bei Erwachsenen und < 90% bei Jugendlichen) vorliegen. Patienten der Studie 1 hatten eine reguläre Behandlung mit hochdosierten inhalierten Kortikosteroiden ICS, Patienten in Studie 2 eine ICS-Behandlung mit mittlerer oder hoher Dosis in Kombination mit lang wirksamen Beta-Agonisten (LABA). Die mittlere Anzahl von Exazerbationen im vorausgegangenen Jahr war 3 und die durchschnittliche vorhergesagte «vor Bronchodilatation» FEV1 war 57,5%. Die Patienten wurden nach geographischer Lage, Alter und Eosinophilenanzahl im peripheren Blut stratifiziert (≥300 Zellen/μl oder < 300 Zellen/μl).
An der Placebo-kontrollierten Studie 3 (ZONDA) zur Reduktion oraler Kortikosteroide (OCS) nahmen insgesamt 220 Asthmapatienten (61% Frauen; Durchschnittsalter 51 Jahre) teil, 73 davon wurden mit der beantragten Dosis behandelt. In die Studie wurden Patienten eingeschlossen, die täglich mit OCS (7,5–40 mg pro Tag) behandelt wurden und zusätzlich regelmässig hochdosierte ICS und LABA mit oder ohne zusätzliche Bedarfsmedikamente einnahmen, um die Asthmakontrolle aufrechtzuerhalten. Die Studie umfasste einen 8-wöchigen Run-in-Zeitraum, in dem die OCS auf die minimal wirksame Dosis titriert wurden, ohne die Asthmakontrolle zu verlieren. Die mittlere OCS-Dosis bei Baseline lag bei allen Behandlungsgruppen auf ähnlichem Niveau. Die Patienten mussten eine Eosinophilenanzahl im peripheren Blut von mindestens 150 Zellen/μl sowie mindestens eine Exazerbation in den vergangenen 12 Monaten aufweisen. Die mittlere OCS-Dosis bei Baseline lag bei 10 mg (Bereich 8–40 mg) bei allen 3 Behandlungsgruppen. An der einarmigen Studie 6 (PONENTE) zur Reduktion der OCS nahmen insgesamt 598 Patienten (64% weiblich; Durchschnittsalter 53 Jahre) teil, die alle mit der beantragten Dosis behandelt wurden. Die mittlere OCS-Dosis bei Baseline betrug 10 mg (Bereich 5–60 mg).
Während zwei Dosisschemata in den Studien 1, 2 und 3 untersucht wurden, ist das empfohlene Dosisschema von Fasenra alle 4 Wochen für die ersten 3 Dosen und danach alle 8 Wochen (siehe «Dosierung/Anwendung»).
Exazerbationen
Den primären Endpunkt für Studie 1 (SIROCCO) und Studie 2 (CALIMA) stellte die jährliche Häufigkeit klinisch signifikanter Asthma-Exazerbationen bei solchen Patienten dar, die bei Baseline einen Eosinophilenwert im Blut von mindestens 300 Zellen/μl aufwiesen und die hochdosierte ICS und LABA inhalierten. Eine klinisch signifikante Asthma-Exazerbation wurde als eine Verschlechterung des Asthmas definiert, bei der aufgrund des Asthmas mindestens 3 Tage lang orale/systemische Kortikosteroide verwendet werden mussten, und/oder eine stationäre Aufnahme erfolgte, bei der eine Behandlung mit oralen/systemischen Kortikosteroiden notwendig war und/oder die Notaufnahme aufgesucht werden musste. Für Patienten, die orale Kortikosteroide als Erhaltungstherapie einnahmen, wurde eine klinisch signifikante Asthma-Exazerbation wie folgt definiert: Kurzzeitige Erhöhung der vorher stabilen oralen/systemischen Kortikosteroide über mindestens 3 Tage oder Gabe einer einzelnen Depot-Injektion eines Kortikosteroids.
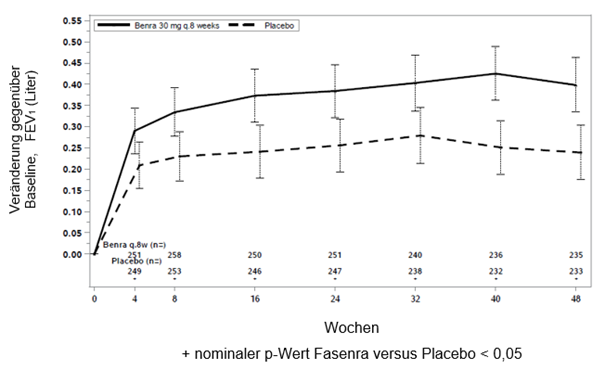
In beiden Studien 1 und 2 zeigten Patienten mit mindestens 300 Zellen/μl Eosinophile im Blut unter Fasenra eine signifikante Verringerung der jährlichen Exazerbationsrate verglichen mit Placebo (Tabelle 1). Änderung der durchschnittlichen FEV1 gegenüber der Baseline wurde in beiden Studien gemessen und hat zumindest numerische Vorteile zu jedem Zeitpunkt ab 4 Wochen gezeigt (siehe Abbildung 1). Die Wirkung wurde bis zum Ende der gesamten Behandlung, wie in Tabelle 1 gezeigt, erhalten.
Tabelle 1. Ergebnisse für die jährliche Exazerbationsrate und Lungenfunktion am Ende der Behandlung von Studie 1 (SIROCCO) und 2 (CALIMA) gegen die Anzahl der Eosinophilen
|
|
Studie 1 |
Studie 2 | ||
|
Fasenra |
Placebo |
Fasenra |
Placebo | |
|
Anzahl der Eosinophile im Blut ≥300 Zellen/μla |
n=267 |
n=267 |
n=239 |
n=248 |
|
Klinisch signifikante Exazerbationen | ||||
|
Rate |
0,74 |
1,52 |
0,73 |
1,01 |
|
Differenz |
-0,78 |
-0,29 | ||
|
Rate ratio (95% KI) |
0,49 (0,37; 0,64) |
0,72 (0,54; 0,95) | ||
|
p-Wert |
< 0,001 |
0,019 | ||
|
FEV1 vor Bronchodilatation (Liter) | ||||
|
Durchschnitt Baseline |
1,660 |
1,654 |
1,758 |
1,815 |
|
Verbesserung gegenüber der Baseline |
0,398 |
0,239 |
0,330 |
0,215 |
|
Differenz (95% KI) |
0,159 (0,068; 0,249) |
0,116 (0,028; 0,204) | ||
|
p-Wert |
0,001 |
0,010 | ||
a Intent-to-Treat (ITT)-Population (Patienten unter Behandlung von hochdosierten ICS und Eosinophilen im Blut ≥300 Zellen/μl).
Eine kombinierte Analyse der Studien 1 und 2 zeigte eine stärkere Verminderung der Exazerbationsrate mit zunehmender Anzahl Eosinophilen im Blut bei Baseline. Ebenfalls hat eine kombinierte Analyse eine grössere Verbesserung von FEV1 bei Patienten mit zunehmender Anzahl Eosinophiler im Blut gezeigt.
Für Studie 1 (SIROCCO) war die Rate der Exazerbationen, die eine stationäre Aufnahmen und/oder einen Besuch der Notaufnahme erforderte, für Patienten unter Fasenra im Vergleich zu Placebo 0,09 versus 0,25 (Rate ratio 0,37, 95% KI: 0,20, 0,67, p ≤0,001). Diese Rate war für Studie 2 (CALIMA) 0,12 versus 0,10 (Rate ratio 1,23; 95% KI: 0,64; 2,35; p=0,538). Studie 2 zeigte diesbezüglich numerisch keine Vorteile für Fasenra. Allerdings war die Ereignisrate zu gering, um Schlussfolgerungen zur Wirkung von Fasenra auf schwere Exazerbationen ziehen zu können, die stationäre Aufnahmen und/oder Besuche der Notaufnahme erforderten.
Abbildung 1. Mittlere Änderung gegenüber der Baseline FEV1 vor Bronchodilatation (Liter), Studie 1 (SIROCCO) und 2 (CALIMA)
Studie 1
Studie 2
Die Anzahl an Patienten, bei denen mindestens eine Exazerbation auftrat, war unter Fasenra niedriger im Vergleich zu Placebo (35% vs 51% in Studie 1 und 40% vs 51% in Studie 2), und die Zeit bis zur 1. Exazerbation war länger für Fasenra in beiden Studien (Hazard ratio 0,60; 95% KI: 0,46; 0,78 und Hazard ratio 0,73; 95% KI: 0,55; 0,95 entsprechend in Studie 1 und 2).
Zusätzlich wurde am Ende der Behandlung eine Verbesserung der mittleren Änderung des morgendlichen und abendlichen Peak Expiratory Flow (PEF) gegenüber der Baseline bei Patienten unter Fasenra im Vergleich zu Placebo beobachtet.
In beiden Studien 1 und 2 trat bei Patienten, die Fasenra erhalten hatten, eine statistisch signifikante Reduktion an Asthmasymptomen auf (Gesamt Asthma-Score) in Vergleich zu Patienten, die Placebo erhalten hatten. Eine konsistente Verbesserung in Bezug auf Fasenra wurde im Zuge des Asthmakontrollfragebogens (Asthma Control Questionnaire-6: ACQ-6) und des standardisierten Asthmafragebogens zur Lebensqualität für Personen ab 12 Jahren (Standardised Asthma Quality of Life Questionnaire for 12 Years and Older: AQLQ(S)+12) erhalten (Tabelle 2).
Tabelle 2. Behandlungsunterschied als mittlere Änderung gegenüber der Baseline des Asthmasymptom-Score, ACQ-6 und AQLQ(s)+12 am Ende der Behandlunga
|
Wirksamkeitsvariable |
Studie 1 |
Studie 2 | ||
|
Fasenra |
Placebo |
Fasenra |
Placebo | |
|
Gesamter Asthmasymptom-Scoreb | ||||
|
Durchschnittliche Baseline |
2,68 |
2,74 |
2,76 |
2,71 |
|
Verbesserung gegenüber Baselineb |
-1,30 |
-1,04 |
-1,40 |
-1,16 |
|
Differenz (95% KI) |
-0,25 (-0,45; -0,06) |
-0,23 (-0,43; -0,04) | ||
|
p-Wert |
0,012 |
0,019 | ||
|
ACQ-6 | ||||
|
Durchschnittliche Baseline |
2,81 |
2,90 |
2,80 |
2,75 |
|
Verbesserung gegenüber Baseline |
-1,46 |
-1,17 |
-1,44 |
-1,19 |
|
Differenz (95% KI) |
-0,29 (-0,48; -0,10) |
-0,25 (-0,44; -0,07) | ||
|
AQLQ(S)+12 | ||||
|
Durchschnittliche Baseline |
3,93 |
3,87 |
3,87 |
3,93 |
|
Verbesserung gegenüber Baseline |
1,56 |
1,26 |
1,56 |
1,31 |
|
Differenz (95% KI) |
0,30 (0,10, 0,50) |
0,24 (0,04; 0,45) | ||
a Patientenanzahl (n) variiert leicht auf Grund der Patientenanzahl, für welche Daten für die jeweilige Variable vorhanden waren. Die angegebenen Resultate basieren auf den letzten verfügbaren Daten für jede Variable.
b Asthmasymptom-Score: Gesamt-Score von 0 (niedrigste) bis 6 (höchste); Tag- und Nacht-Zeit Asthmasymptom-Score von 0 (niedrigste) bis 3 (höchste) Symptome. Individuelle Tag- und Nacht-Zeit Scores waren ähnlich.
Untergruppen Analysen nach Exazerbationsvorgeschichte
Eine Analyse der Subgruppen aus Studie 1 und 2 identifizierte eine höhere Exazerbationsrate in der Vorgeschichte als potentiellen Prädiktor für ein verbessertes Ansprechen auf die Behandlung mit Benralizumab. Die Exazerbationsrate kann alleine oder kombiniert mit einer höheren Baseline-Anzahl von Eosinophilen im Blut zur Identifikation von Patienten herangezogen werden, welche ein besseres Ansprechen auf die Benralizumab Behandlung erreichen könnten.
In beiden Studien zeigten Patienten, die innerhalb von 12 Monaten vor der Fasenra-Randomisierung 3 oder mehr Exazerbationen hatten, eine grössere Senkung der Exazerbationsrate, als Patienten mit weniger als 3 vorherigen Exazerbationen (siehe Tabelle 3).
Tabelle 3: Exazerbationsrate und Lungenfunktion (FEV1) am Behandlungsende nach Anzahl der Exazerbationen im vorherigen Jahr, Studie 1 (SIROCCO) und 2 (CALIMA) (Intent-to-Treat Population)
|
|
Studie 1 |
Studie 2 | ||
|
|
Fasenra (N=267) |
Placebo (N=267) |
Fasenra (N=239) |
Placebo (N=248) |
|
Baseline mit 2 Exazerbationen | ||||
|
n |
164 |
149 |
144 |
151 |
|
Exazerbationsrate |
0,57 |
1,04 |
0,63 |
0,62 |
|
Differenz |
-0,47 |
0,01 | ||
|
Inzidenzrate |
0,55 |
1,01 | ||
|
Durchschnittliche Veränderung FEV1 vor Bronchodilatation |
0,343 |
0,230 |
0,266 |
0,236 |
|
Inzidenzrate |
0,113 |
0,029 | ||
|
Baseline mit mehr als 3 Exazerbationen | ||||
|
n |
103 |
118 |
95 |
97 |
|
Exazerbationsrate |
0,95 |
2,23 |
0,82 |
1,65 |
|
Differenz |
-1,28 |
-0,84 | ||
|
Inzidenzrate |
0,43 |
0,49 | ||
|
Durchschnittliche Veränderung FEV1 vor Bronchodilatation |
0,486 |
0,251 |
0,440 |
0,174 |
|
Inzidenzrate |
0,235 |
0,265 | ||
Hinsichtlich der Asthmasymptome wiesen Patienten mit einer Vorgeschichte von 3 oder mehr Exazerbationen in den 12 Monaten vor der Randomisierung am Ende der Behandlung mit Fasenra in Studie 1 bzw. 2 mittlere Score-Differenzen von -0,32 und -0,41 im Vergleich zur Baseline auf (Studie 1 95% KI: -0,62, -0,01; Studie 2 95% KI: -0,73, -0,09). Patienten mit einer Vorgeschichte von 2 Exazerbationen in den 12 Monaten vor der Randomisierung zeigten am Ende der Behandlung mit Fasenra in Studie 1 bzw. 2 mittlere Punktzahl-Differenzen hinsichtlich der Asthmasymptome von -0,22 und -0,12 im Vergleich zur Baseline (Studie 1: 95% KI: -0,49; -0,04; Studie 2: 95% KI: -0,37; -0,13).
Studien zur Verringerung der Oralen Kortikosteroid (OCS)-Anwendung
Die Studien ZONDA (Studie 3), eine Placebo-kontrollierte Studie, und PONENTE (Studie 6), eine offene Studie, beurteilten die Wirkung von Fasenra in Bezug auf die Verringerung der Anwendung von oralen OCS als Erhaltungstherapie.
In Studie 3 war der primäre Endpunkt die prozentuale Verringerung der OCS-Dosis bei Studienabschluss in den Wochen 24–28 gegenüber der Baseline, bei gleichzeitig fortgesetzter Asthmakontrolle. Im Vergleich zu Placebo konnten die mit Fasenra behandelten Patienten die tägliche Erhaltungsdosis oraler Kortikosteroide bei gleichzeitiger Erhaltung der Asthmakontrolle stärker senken. Eine Verringerung der OCS-Dosis von 50% oder mehr wurde bei 48 Patienten (66%) beobachtet, welche Fasenra erhielten, verglichen zu 28 Patienten (37%), welche Placebo erhielten. Die Tabelle 4 fasst die Ergebnisse der Studie 3 zusammen.
Tabelle 4. Wirkung von Fasenra auf die Verringerung der OCS-Dosis, Studie 3 (ZONDA)
|
|
Fasenra |
Placebo |
|
Wilcoxon-rank sum test (primäre Analysemethode) | ||
|
Mediane prozentuale Verringerung der täglichen OCS-Dosis gegenüber Baseline (95% KI) |
75 (60; 88) |
25 (0; 33) |
|
Wilcoxon-rank sum test, p-Wert |
< 0,001 |
|
|
Proportional Odds Model (Empfindlichkeitsanalyse) | ||
|
Prozentuale Reduktion der OCS im Vergleich zur Baseline bis Woche 28 | ||
|
≥90% |
27 (37%) |
9 (12%) |
|
≥75% |
37 (51%) |
15 (20%) |
|
≥50% |
48 (66%) |
28 (37%) |
|
> 0% |
58 (79%) |
40 (53%) |
|
Keine Änderung oder keine Verringerung an OCS |
15 (21%) |
35 (47%) |
|
Odds Ratio (95% KI) |
4,12 (2,22; 7,63) |
|
|
Reduktion der täglichen OCS-Dosis auf 0 mg/Tag* |
22 (52%) |
8 (19%) |
|
Odds Ratio (95% KI) |
4,19 (1,58; 11,12) |
|
|
Reduktion der täglichen OCS-Dosis auf ≤5 mg/Tag |
43 (59%) |
25 (33%) |
|
Odds Ratio (95% KI) |
2,74 (1,41; 5,31) |
|
|
Exazerbationsrate |
0,54 |
1,83 |
|
Rate ratio (95% KI) |
0,30 (0,17; 0,53) |
|
|
Exazerbationsrate, die stationäre Aufnahmen/Besuche der Notaufnahme erforderten |
0,02 |
0,32 |
|
Rate ratio (95% KI) |
0,07 (0,01; 0,63) |
|
* Nur Patienten mit optimierter Baseline OCS-Dosis von 12,5 mg oder weniger konnten eine 100% Reduktion der OCS-Dosis während der Studie erreichen.
Zusätzlich wurden in Studie 3 die Lungenfunktion, der Asthmasymptomscore, der ACQ-6 und der AQLQ(S)+12 untersucht. Die Resultate waren vergleichbar zu den Resultaten aus Studie 1 und 2.
In die Studie 6 wurden 598 erwachsene Patienten mit schwerem Asthma (Eosinophilenzahl im Blut von ≥150 Zellen/μl bei Aufnahme in die Studie oder ≥300 Zellen/μl in den letzten 12 Monaten, wenn die Eosinophilenzahl im Blut bei Aufnahme in die Studie < 150 Zellen/μl betrug) aufgenommen, die von oralen Kortikosteroiden abhängig waren. Primäre Endpunkte waren der Anteil an Patienten, die unter Aufrechterhaltung der Asthmakontrolle die OCS absetzen konnten, und der Anteil an Patienten, die unter Aufrechterhaltung der Asthmakontrolle und unter Berücksichtigung der Nebennierenfunktion eine finale OCS-Dosis von 5 mg oder weniger erreichten. Der Anteil an Patienten, die die OCS-Erhaltungstherapie absetzen konnten, betrug 62,9%. Der Anteil an Patienten, die eine finale OCS-Dosis von 5 mg oder weniger erreichten (unter Aufrechterhaltung der Asthmakontrolle und ohne Einschränkung der Nebennierenfunktion), betrug 81,9%. Die Wirkungen auf die OCS-Reduktion waren unabhängig von der Eosinophilenzahl im Blut bei Aufnahme in die Studie (einschliesslich Patienten mit einer Eosinophilenzahl im Blut von < 150 Zellen/μl) vergleichbar und wurden über einen zusätzlichen Zeitraum von 24 bis 32 Wochen aufrechterhalten. Die jährliche Exazerbationsrate war in Studie 6 vergleichbar mit der in den vorhergehenden Studien berichteten Rate.
Langzeit-Extensionsstudien
Die Langzeitsicherheit von Fasenra wurden im Rahmen einer doppelblinden, randomisierten, 56-wöchigen Extensionsstudie der Phase 3 mit Parallelgruppen beurteilt (Studie 4, BORA). Die Langzeitsicherheit von Fasenra wurde zudem in der offenen Sicherheits-Extensionsstudie MELTEMI (Studie 5) untersucht (siehe «Unerwünschte Wirkungen»).
In Studie 4 (BORA) wurden 2123 erwachsene und jugendliche Patienten (ab 12 Jahren) aus den Studien 1 (SIROCCO), 2 (CALIMA) und 3 (ZONDA) aufgenommen. Davon wurden 842 Patienten mit Fasenra mit der genehmigten Dosierung behandelt.
Studie 4 (BORA) diente primär der Untersuchung der Langzeitsicherheit und -verträglichkeit von Fasenra. Zudem wurde der weitere Verlauf der jährlichen Exazerbationsrate, Lungenfunktion, ACQ-6, AQLQ(S)+12 sowie der OCS-Dosis beobachtet. Dabei wurde die in den beiden vorherigen Studien evaluierten Dosierungsschemata angewendet.
Die in den vorherigen Studien 1 und 2 im Vergleich zu Placebo festgestellte geringere Exazerbationsrate und OCS-Dosis war auch im zweiten Jahr unter Anwendung des genehmigten Dosierungsschemas zu verzeichnen. Die in den vorherigen Studien 1 und 2 festgestellten Verbesserungen bei der Lungenfunktion, ACQ-6 und AQLQ(S)+12 blieben ebenfalls im zweiten Jahr unter Anwendung des genehmigten Dosierungsschemas erhalten.
In Studie 5 wurde die Langzeitsicherheit bei 226 Patienten untersucht, die mindestens eine Dosis von Fasenra im empfohlenen Dosierungsschema erhielten. Die jährliche Exazerbationsrate in Studie 5 (0,47) war vergleichbar mit der in den Studien 1 (0,74), 2 (0,73) und 4 (0,46).
Immunogenität
Während des Placebo-kontrollierten 48- bis 56-wöchigen Phase 3-Behandlungszeitraums, in dem die Patienten mit Fasenra im empfohlenen Dosisschema behandelt wurden, entwickelten 107 von 809 Patienten (13%) eine behandlungsbedingte antimedikamentöse Antikörperreaktion (ADA). Die Mehrheit der ADA-positiven Studienteilnehmer wies in-vitro-neutralisierende Antikörper auf. Im Vergleich zu den Antikörper-negativen Studienteilnehmern wurden Patienten mit hohem Anti-Benralizumab-Antikörpertiter in Verbindung gebracht mit verringerten Benralizumab-Konzentrationen im Serum und erhöhten Eosinophilenwerten im Blut. Bis zum gegenwärtigen Zeitpunkt wurde kein Zusammenhang zwischen Anti-Benralizumab-Antikörpern und Veränderungen hinsichtlich Wirksamkeit oder Sicherheit beobachtet.
Nach dem zweiten Behandlungsjahr traten unter der Fasenra-Behandlung mit dem empfohlenen Dosisschema bei weiteren 18 von 510 (4%) Patienten mit vorheriger Fasenra Behandlung und bei 29 von 279 (10,4%) Patienten mit vorheriger Placebo-Behandlung Antikörper auf. Die Gesamt-Prävalenz von Antikörpern (unabhängig von der Vorbehandlung) betrug 11,7%, diejenige von neutralisierenden Antikörper 9,5%.
Die Titer bei den Patienten, die in den vorangegangenen Studien ADA-positiv gewesen waren, blieben durchschnittlich im ersten Jahr stabil und waren im zweiten Behandlungsjahr tendenziell eher rückläufig. Dennoch waren bei einigen Patienten anhaltend hohe Titer zu verzeichnen. Die berichteten Nebenwirkungen bei ADA-positiven Patienten, einschliesslich der Patienten mit anhaltend hohen ADA-Titern, glichen denen von ADA-negativen Patienten und entsprachen der Grunderkrankung der untersuchten Patientengruppe.
In Einklang mit den vorangegangenen Studien wurde keine Assoziation zwischen Anti-Benralizumab-Antikörpern und Wirksamkeit oder Sicherheit gefunden.
Die Daten spiegeln den Anteil an Patienten wider, deren Testresultate positiv für Benralizumab-Antikörper in spezifischen Assays ausfielen. Die beobachtete Inzidenz der Antikörper-Antwort ist stark von verschiedenen Faktoren abhängig, wie der Assay Sensitivität und Spezifität, der Methode des Assays, dem Umgang mit den Proben, dem Timing der Probenabnahme, Begleitmedikation und einer schon vorhandenen Krankheit. Aus diesen Gründen kann der Vergleich der Inzidenz von Benralizumab-Antikörpern mit der Inzidenz von Antikörpern gegen andere Produkte irreführend sein.
Fertilität
Es wurden keine Fertilitätsstudien an Menschen durchgeführt. In tierexperimentellen Studien hat die Behandlung mit Benralizumab keine negative Auswirkung auf die Fertilität (siehe «Präklinische Daten»).
Pharmakokinetik
Die Pharmakokinetik von Benralizumab war bei Patienten mit Asthma nach der subkutanen Verabreichung über einen Dosisbereich von 2 bis 200 mg dosisproportional.
Absorption
Nach der subkutanen Verabreichung an Patienten mit Asthma lag die Absorptions-Halbwertszeit bei 3,5 Tagen. Basierend auf der pharmakokinetischen Analyse der Population lag die geschätzte, absolute Bioverfügbarkeit bei etwa 59% und es gab keine klinisch relevanten Unterschiede bezüglich der relativen Bioverfügbarkeit bei einer Administration ins Abdomen, in den Oberschenkel oder in den Oberarm.
Distribution
Basierend auf der pharmakokinetischen Analyse der Population wird für eine 70 kg schwere Person das zentrale und periphere Verteilungsvolumen von Benralizumab auf 3,1 Liter bzw. 2,5 Liter geschätzt.
Biotransformation
Benralizumab ist ein humanisierter IgG1 monoklonaler Antikörper, welcher durch proteolytische Enzyme abgebaut wird, welche im ganzen Körper und nicht nur in der Leber vorhanden sind.
Elimination
Die Populations-Pharmakokinetik-Analyse von Benralizumab spricht für eine lineare Pharmakokinetik ohne Zielrezeptor-vermittelten Clearance-Pfad. Die geschätzte systemische Clearance (CL) für Benralizumab lag bei 0,29 Liter pro Tag. Nach der subkutanen Verabreichung lag die Eliminations-Halbwertzeit bei etwa 15,5 Tagen.
Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine formalen Studien zur Erfassung von Wechselwirkungen mit anderen Arzneimitteln durchgeführt.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Zum Einfluss einer Leberfunktionsbeeinträchtigung auf die Pharmakokinetik von Benralizumab wurden keine klinischen Studien durchgeführt. Da IgG-monoklonale Antikörper nicht hauptsächlich über die Leber abgebaut werden, ist keine Beeinflussung der Clearance von Benralizumab durch eine Leberfunktionsstörung zu erwarten. Basierend auf populationspharmakokinetischen Analysen hatten die bei Baseline gemessenen Leberfunktionsbiomarker (ALT, AST und Bilirubin) keinen klinisch relevanten Effekt auf die Clearance von Benralizumab.
Nierenfunktionsstörungen
Zum Einfluss einer Nierenfunktionsbeeinträchtigung auf die Pharmakokinetik von Benralizumab wurden keine formalen klinischen Studien durchgeführt. Basierend auf populationspharmakokinetischen Analysen war die Benralizumab Clearance in Patienten mit einer Kreatinin Clearance zwischen 30 und 80 ml/min und in Patienten mit normaler Nierenfunktion vergleichbar. Es sind limitierte Daten verfügbar für Patienten mit einer Kreatinin Clearance unter 30 ml/min, aber Benralizumab wird nicht durch die Niere ausgeschieden.
Ältere Patienten
Die populationspharmakokinetische Analyse ergab keine Hinweise auf einen Einfluss des Alters auf die Clearance von Benralizumab.
Geschlecht
Eine populationspharmakokinetische Analyse ergab, dass das Geschlecht keinen signifikanten Einfluss auf die Clearance von Benralizumab hat.
Präklinische Daten
Sicherheitspharmakologie
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie oder wiederholten Dosistoxizitätsstudien an Affen lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Da es sich bei Benralizumab um einen monoklonalen Antikörper handelt, wurden keine Genotoxizitäts- oder Kanzerogenitätsstudien durchgeführt.
Eine Abnahme der Eosiniphilenzahl im peripheren Blut und im Knochenmark wurde in der Mehrheit der Cynomolgus-Affen nach intravenöser und subkutaner Verarbeichung von Benralizumab beobachtet. Das fehlende Ansprechen mancher Affen konnte nicht abschliessend geklärt werden. Die Reduktion der Eosinophilenzahl bedingte keine toxikologisch relevanten Befunde.
Reproduktionstoxizität
In einer Studie zur prä- und postnatalen Entwicklung bei Cynomolgus-Affen zeigte eine Gabe von 10 oder 30 mg/kg Benralizumab keine Auswirkung auf die maternale oder die embryofetale bzw. postnatale Entwicklung. Bei Affen, die während der Trächtigkeit exponiert waren, wurde bei den Nachkommen eine Reduktion der Eosinophilen gezeigt.
Fertilität
Es gab keine Auswirkung auf die Fertilität von männlichen und weiblichen Cynomolgus-Affen in den Studien mit wiederholter Verabreichung von bis zu 30 mg/kg Benralizumab.
Sonstige Hinweise
Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Fasenra kann bei bis zu 25°C über einen Zeitraum von maximal 14 Tagen aufbewahrt werden. Nach der Entnahme aus dem Kühlschrank muss Fasenra innerhalb von 14 Tagen angewendet oder verworfen werden. Keiner Hitze aussetzen. Nicht einfrieren. Nicht schütteln.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Nicht schütteln. Falls eingefroren nicht mehr verwenden.
Vor der Anwendung Fasenra der Umgebungstemperatur anpassen indem es im Umkarton bei Umgebungstemperatur (bis zu 25°C) liegen gelassen wird. Dies dauert in der Regel 30 Minuten. Untersuchen Sie Fasenra vor der Anwendung visuell auf Partikel oder Verfärbungen.
Fasenra ist klar, farblos bis gelb und kann lichtdurchlässige oder weisse bis cremefarbene Partikel enthalten. Verwenden Sie Fasenra nicht, wenn die Flüssigkeit trübe oder verfärbt ist oder wenn sie grosse Partikel oder fremde Bestandteile enthält.
Die Packungsbeilage enthält eine ausführliche Gebrauchsanweisung.
Zulassungsnummer
67581 (Swissmedic)
Packungen
Packung mit 1 Fasenra Pen zu 30 mg / 1 ml. [B]
Zulassungsinhaberin
AstraZeneca AG, 6340 Baar
Stand der Information
Dezember 2022